Insulinoma
greg
Glucagonoma
grg
Phechromocytoma
dg
Cushing syndrome
dqwdqd
Cushing disease
dwqd
Acromegaly
dqwdq
Diabetes 1: Symptoms, prevalence, genetics, pathogenesis, complications, and treatment
What is insulinitis?
Antibodies (types)
Lack of insulin which causes hyperglycemia.
Classical symptoms: polyuria, polydipsia, polyphagia, and weight loss. Additional symptoms may include blurry vision, feeling tired, and poor healing.
Prevelance: Affects people under 30 (and in late late stages in diabets 2). 5-10 % of people with diabetes
Genetics: HLA genes (chromosome 6) - 90% of all with diabetes 1 have either one or two of these below, which makes them succeptible to immune mediated destruction.
- HLA-DR3-DQ2
- HLA-DR4-DQ8
Trigger: Exposure to enteroviruses such as Coxsackie B4 have been associated with the initiation of the destruction of pancreatic beta-cells. Many different teoires.
Insulinitis: Insulitis is an inflammation of the beta cells in the islets of Langerhans. CD8+ cytotoxic T-cells and macrophages in the early stage and B-cells in later stages. Spesific antibodies againgst beta-cells can be detected. TNF-a, INF-a, and INF-g are participate.
Antibodies:
- Islet cell antibodies (ICA), can be detected years before onset and are present in 60-80% of all newly diagnosed cases.
- Insulin antibodies (IAA), 100% detectable in T1D under the age of 5 and 20% of cases ocer 15 years.
- Glutamic acid decarboxylase antibodies (GADA), detectable in 80% of patients years beore onset.
The appearance of diabetes-related autoantibodies has been shown to be able to predict the appearance of diabetes type 1 before any hyperglycemia arises, the main ones being islet cell autoantibodies, insulin autoantibodies, glutamic acid decarboxylase (GAD).
Diabetes 2: Prevalence, symptoms, genetics, environmental factors (risk factors), monogenic inheritance, insunlin resistance, obesity and “thrifty” gene
Prevalence: 90% of all diabetics. Starts slowly and affetcts primarly people over 40 year.
Symptoms:
Genetics and environmental factors (risk factors)
- Strong genetic predisposition (family history - 2-4x increased risk and 88% concordance in twins!)
- Polygenic disorder - polymorphisms in;
- IRS1, IRS2, B3-adrenergic receptos ++
- Metabolic syndrome!
- Low birth weight
- Sedentary lifestyle
Monogenic inheritance: Insulin receptor mutation, maternally inherited diabetes and deafness (MIDD), wolfram syndrome, MODY (7 types).
Insulin resistance: Pathological condition in which cells fail to respond normally to the hormone insulin. Beta cells in the pancreas subsequently increase their production of insulin, further contributing to a high blood insulin level.
“Thrifty” gene hypotisis: process of natural selection. Genes which predispose to diabetes (called ‘thrifty genes’) were historically advantageous, but they became detrimental in the modern world. Thrifty genes are genes which enable individuals to efficiently collect and process food to deposit fat during periods of food abundance in order to provide for periods of food shortage (feast and famine).
Addison`s disease
dwq
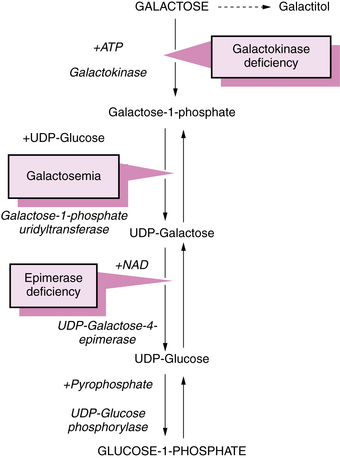
Galactose metabolism deficiencies
Three types 1: Classic galactosemia - galactose 1 phosphate uridyltransferase (fatique, vometing, hepatomegaly and jaundice, diabetes, cataract and developmental delay) 2: Galactokinase deficiency - Galactokinase (cataract) 3: Galactose epimerase deficiency - UDP-galactose epimerase(same as type 1)
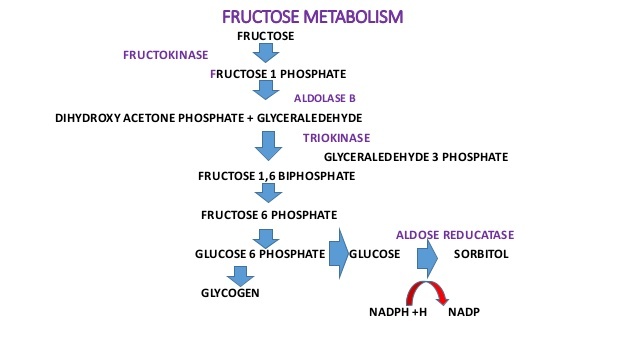
Fructose metabolism deficiencies
Two types 1: Fructouria - fructokinase (fructose in blood and urine) 2: Herediatary fructose intolerance - Aldolase B HFI - Asymptomatic until they ingest fructose, sucrose, or sorbitol. - Accumulation of fructose-1-phosphate which have downstream effects on gluconeogenesis and regeneration of ATP. - Symptoms of HFI include vomiting, hypoglycemia, jaundice, hemorrhage, hepatomegaly, hyperuricemia and potentially kidney failure.
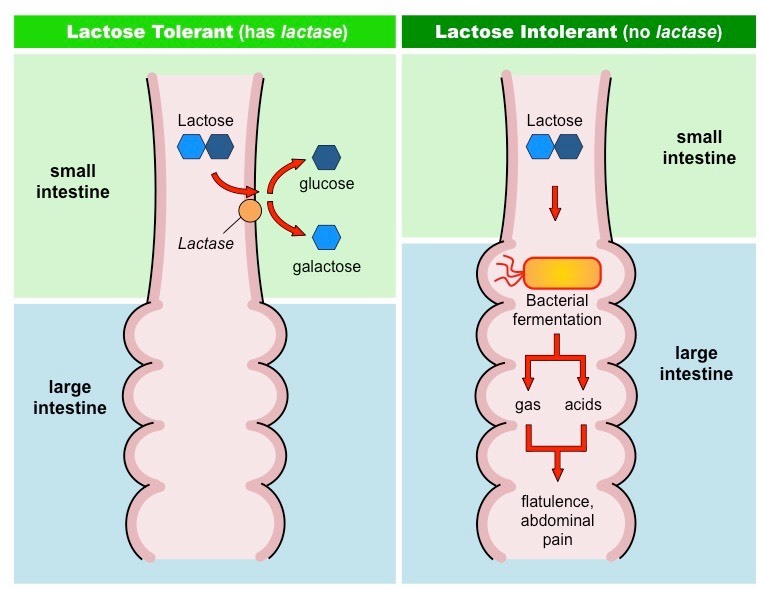
Lactose intolerance: What is missing, symptoms, and what should the patient not consume?
- Intestinal lactase deficiency
- Abdominal cramps and diarrhea
- Bacterial fermentation: CO2, CH4 and H2
- Increased osmotic gradint cause diarrhea
- Avoid milk, cheese and other diary products
Glycogen storage diseases
Type 1 - Von Gierke
Type 2 - Pompe
Type 3: Cori’s disease or Forbes’ disease
Type 4: Andersen disease
Type 5: McArdle
Type 6: Hers’ disease
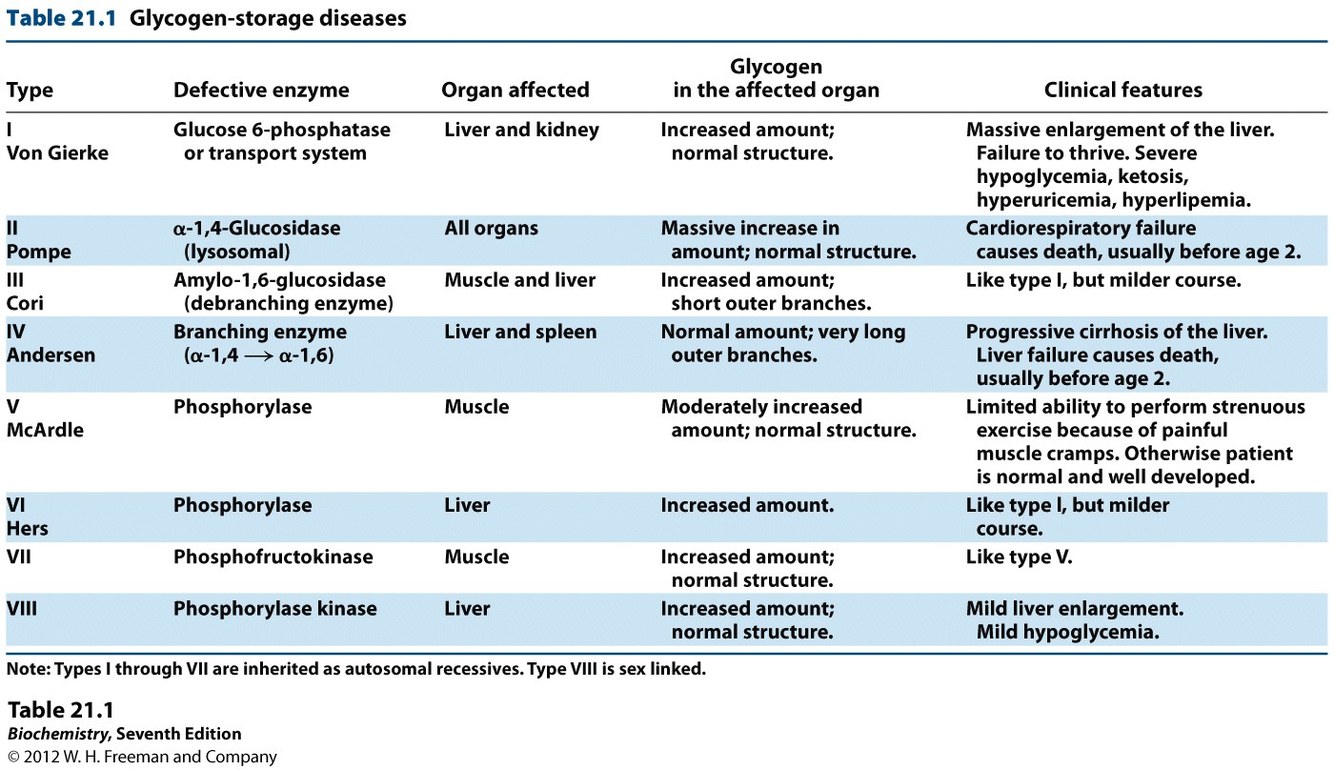
Pathomechanism of diabetic ketoacidosis and hyperosmolar hyperglycemic state: What is the major differance? symptoms and treatment.
What are the 3 ketone bodies? which one is the only “true” ketoacid?
HHS: complication of diabetes mellitus (predominantly type 2) in which high blood sugars cause severe dehydration (polyuria w/osmotic diuresis), increases in osmolarity (relative concentration of solute) and a high risk of complications, coma and death.
- Ketosis is absent because the presence of some insulin inhibits hormone-sensitive lipase mediated fat tissue breakdown.
- A relative insulin deficiency leads to a serum glucose that is usually higher than 33 mmol/L (600 mg/dL), and a resulting serum osmolarity that is greater than 320 mOsm.
- Treatment: intravenous fluids, reduction of the blood sugar levels with insulin, and management of any underlying conditions that might have precipitated the illness, such as an acute infection.
DKA: often (but not exclusively!) encountered in people with type 1 diabetes; they are differentiated with measurement of ketone bodies, organic molecules that are the underlying driver for DKA but are usually not detectable in HHS.
- Kussmaul breathing, fruity odor (acetone), polyuria (osmotic diuresis), nausea and vomiting, abdominal pain, and coma.
- DKA is typically diagnosed when testing finds high blood sugar, low blood pH, and ketoacids in either the blood or urine.
- Treatment with insulin, IV fluids, and monitoring of K+ levels
Ketone bodies: Acetone, beta-hydroxybutyrate, and acetoacetate (true)
Microangiopathy:
- What is it
- where do the usually cause damage
- Common causes
- Pathomechanism
Microangiopathy (i.e. disease of blood vessels) affecting small blood vessels in the body
Common places:
- Retinopathy (diabetic retinopathy)
- Nephropathy (diabetic nephopathy)
- Neuropahty (diabetic nephropathy, especially peripheral neuropathy).
Cause: Diabetes, hypertension ++
In case of diabetes (pathomechanism): Hyperglycemia cause
- Endothelial cells take up more glucose and form more glycoproteins on their surface. “Endothelial dysfunction”.
- Basement membrane grow abnormally thick and becomme weaker; leakage of proteins, slow blood flow, bleedings.
- Pericytes express enzymes which convert glucose into osmologically-active metabolites such as sorbitol leading to hypertonic cell lysis. Reduced capillary intergrity, and leakage of albumin an other proteins over time (particullary in the glumeruli), and proteinuria, and eventually renal failure
Irritable bowel syndrome (IBS): causes, symptoms, risk factors, treatment, pathology.
Symptoms: Reccurent abdoninal pain, and abnormal bowel motility which causes constipation and/or diarrhea.
Causes:“Functional disorder”. Viceral hypersensitivity (e.g. after stretching). Short-chain carbohydrates -> increased osmotic effect -> stretching, smooth muscle spasm and pain. Bacterial fermintation -> gas.
Treatment
- Diet modification (avoid food with short-chain carbohydrates such as apple and coliflower)
- For constipation: soluable fiber, stool softners and osmotic laxatives
- For spasms and pain: Anti-diarrheals and antimuscarinic`s
- Manage: stress, anxienty, and depression
Peripheral vascular disease: Causes, symptoms, physical exam, and treatment)
Causes: coming most often due to atherosclerosis. smoking, diabetes, dyslipedimea and HTN
Symptoms: coming
- Lower limb claudication (pain caused by too little blood flow, usually during exercise), ulcers, colour change. With severe hypoxia there are risk of gangrene.
- Stroke
Physical exam
- Bruit sound on auscultaion over the artery
- Ankle-brachial index: Systolic blood pressure in ankle/systolic pressure in arm = < 0,9
Treatment
- Lifestyle changes: quit smoking, healthy eating and exercise regulary
- Medication: reduce clotting
- Surgary is rarly performed
Liver cirrhosis : Causes, and consequences
Cirrhosis: Damage cells -> fibrosis (scar tissue). Irreversible process.
Stellate cells are a key cell type in fibrosis (they normaly store vitamin A, are “quiescent”, become active after injury and secrete TGF-b and produce collagen. Regenerative nodules.
Causes: Alcohol, virus, or long term inflammation and stressors
Consequences: Fibrosis compress other structures in the portal tirad and cause portal hypertension.
- Decresed production of plasma proteins (albumin)
- Edema and acites
- Bleeding (reduced amount of clotting factors)
- Congestive splenomegaly, portosystemic shunt.
- Hepatorenal failure.
- Hepatoencephalopathy (ammonia, asterixis (tremor), coma.
- Ammonia alter the function in astrocytes, accumulation of glutamine (high glutamine/glutamate-ratio -> reduced amount of neurotransmitters, swelling of the cells)
- Symptoms: confusion, altered mental status, and in severe cases hepatic coma, characterised by an inverted sleep-wake pattern (sleeping by day, being awake at night).
- Estrogen in blood accumulate (spider angiomata, palmar erythema, and gynecomastia)
- Reduced gluconeogenesis and glycogenolysis -> Hypoglycemia
- Also hyperglycemia in the postprandial state (liver can store much glycogen as normal due to loss of parenchyma).
- Decresed uptake of lipoproteins and fatty acids -> hypercholersterolemia and hypertriglyceridemia
- Hepatorenal syndrome: Renal failure without primary abnormalities of the kidney temself. Cause in unknown, however evidence points toward splanchnic casodilation and systemic vasoconstriction, leadning to a severe reduction in renal blood flow, particulary to the cortex.
Incresed unconjugated bilrubin levels in blood (jaundice and icterus). Hypoalbuminemia due to reduction in protein synthesis. Less clotting factors
Alcohol-related liver disease: Alcohol metabolism, pathomechanism of liver damage, stages of liver disease, complications, and treatment
More in tornici`s lecture*
Toxic effect on alcohol depend on gender, body size, and duration:
- Female: 20-40 g/day
- Male: 60-80 g/day
3 pathways:
- Alcohol dehydrogenase
- Catalase (in peroxisome)
- Cytochrome P450 2E1 (CYP2E1)
- Induceble, important in phase 1 biotransformation of some drugs -> increased tolerance in sober state, and increased sensitivity in drunk state
Acetaldehyde are broken down to acetate by acetaldehyde dehydrogenase.
Each dehydrogenase step produce one NADH which have metabolic consequences:
- Reduced gluconeogenesis -> hypoglycemia
- Increased formation of FA
- Inhibition of beta-oxidation
- Lactic acidosis (lactate dehydrogenase is regulated by the NADH/NAD+-ratio).
ROS are formed in the catabolism: damage proteins and DNA
Acetaldehyde can bind to macromolecules and alter its function (acetaldehyde adduct). They can also cause neutrophil infiltrate and damage (alcoholic hepatitis) as these proteins are now considered to be forign to the immune system when expressed on the surface.
Stages in liver disease: Healthy liver <-> Hepatic steatosis (alcoholic fatty liver) -> alcoholic hepatitis -> fibrosis and cirrhosis
- Increased risk for hepatocellular carcinoma. 10-20% of patients with alcoholic cirrhosis.
Complicaitons: coming
- Portocaval shunt
Treatment
- Stop alcohol consumption
- Corticosterioids can supress the immune system
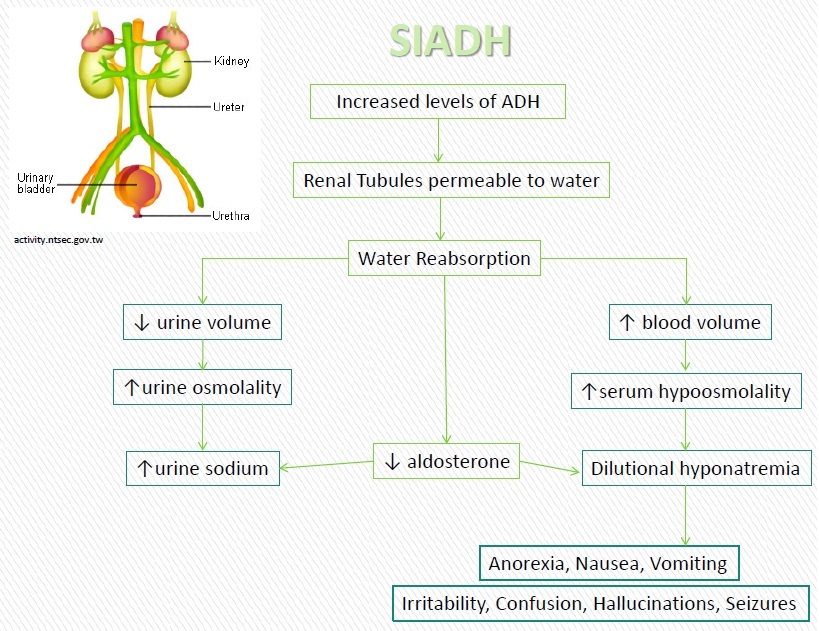
Syndrome of inappropriate secretion of antidiuretic hormone (SIADH): Normal function of ADH, released where, symptoms and pathophysiology
Characterized by excessive release of ADH/vasopressin from the posterior pituitary gland or another source. The increase in blood volume (hypervolemia) often results in dilutional hyponatremia in which the plasma Na+ levels are lowered and total body fluid is increased. Although the sodium level is low, SIADH is brought about by an excess of water rather than a deficit of sodium. SIADH ADH are secreted even when plasma osmolarity is low this futher dilurte the plasma. Aldosterone is not secreted -> low Na+ retention. Plasma sodium osmolarity drop.
Normal function of ADH: Vasoconstriction and fluid retention be translocating AQP2 channels to the luminal surface in the colleting and distal convuluted tubles. More ADH -> more concentrated urine. Lack of ADH results in diluted urine. ADH are secreted in the hypothalamus when plasma osmolarity increase and versa.
Type A: Erratic, independent of plasma osmolarity, high ADH in blood and very concentrated urine.
Type B: Constant release of a moderate amout of ADH
Type C: Release more ADH than normal with the same osmolarity. This condition have a stable, but low plasma Na+
Type D: Normal ADH, but urine osmolarity is still high
Causes:
- Neoplastic: Small-cell carcinoma
- Infections: Pneumonia, meningitis, brain abscess
- Neurological: Subarachnoid hemorrhage
- Iatrogenic: Certain drugs, chemoterapy
- Hypothyroidism, sarcoid
Concequences:
- Cerebral edema due to hyponatremia (can end up in coma)
- Normal BP, serum K+, kidney- and adrenal function
Diagnostics - simultaneous measurment of urine and serum osmolarity
- If serum osmolarity is less than urine osmolarity -> indicate inappropriate excretion of concentrated urine in the presence of very high diluted serum
- Serum Na+ <134 mmol/L, SeOsm <280, urine gravity is >1,005 g/L
Latent autoimmune diabetes of adults: When does it normaly present, which antibodies are usualy present, treatment
Slow onset type 1 diabetes or diabetes type 1.5 is a form of diabetes mellitus type 1 that occurs in adults, often with a slower course of onset.
- Adults with LADA may initially be diagnosed incorrectly as having type 2 diabetes based on their age, particularly if they have risk factors for type 2 diabetes such as a strong family history or obesity.
Develop in patients over 35 years
Autoantibodies:
- Iselet cell antibodies (ICA)
- Glutamic acid decarboxylase antibodies (GADA)
Treatment: Initially with diet and oral antidiabetc drugs, but later with insulin.
Metabolic syndrome
dq
Insulin resistance: Normal action of insulin, peripheral resistance to insulin
Insulin resistance: Pathological condition in which cells fail to respond normally to the hormone insulin. Beta cells in the pancreas subsequently increase their production of insulin, further contributing to a high blood insulin level. Although this type of chronic insulin resistance is harmful, during acute illness it is actually a well-evolved protective mechanism. Recent investigations have revealed that insulin resistance helps to conserve the brain’s glucose supply by preventing muscles from taking up excessive glucose.[2] Insulin resistance should even be strengthened under harsh metabolic conditions such as pregnancy, during which the expanding fetal brain demands more glucose. Insulin resistant cells cannot take in glucose, amino acids and fatty acids. Certain cell types such as fat and muscle cells require insulin to absorb glucose. nsulin resistance in muscle and fat cells reduces glucose uptake (and also local storage of glucose as glycogen and triglycerides, respectively), whereas insulin resistance in liver cells results in reduced glycogen synthesis and storage and also a failure to suppress glucose production and release into the blood. insulin resistance in fat cells reduces the normal effects of insulin on lipids and results in reduced uptake of circulating lipids and increased hydrolysis of stored triglycerides. Increased mobilization of stored lipids in these cells elevates free fatty acids in the blood plasma. Elevated blood fatty-acid concentrations (associated with insulin resistance and diabetes mellitus Type 2), reduced muscle glucose uptake, and increased liver glucose production all contribute to elevated blood glucose levels. If insulin resistance exists, more insulin needs to be secreted by the pancreas. If this compensatory increase does not occur, blood glucose concentrations increase and type 2 diabetes or latent autoimmune diabetes of adults occurs. The presence of insulin leads to a kind of insulin resistance; every time a cell is exposed to insulin, the production of GLUT4 (type four glucose receptors) on the membrane of the cell decreases somewhat.[78] In the presence of a higher than usual level of insulin (generally caused by insulin resistance), this down-regulation acts as a kind of positive feedback, increasing the need for insulin. Exercise reverses this process in muscle tissue,[79] but if it is left unchecked, it may contribute to insulin resistance.
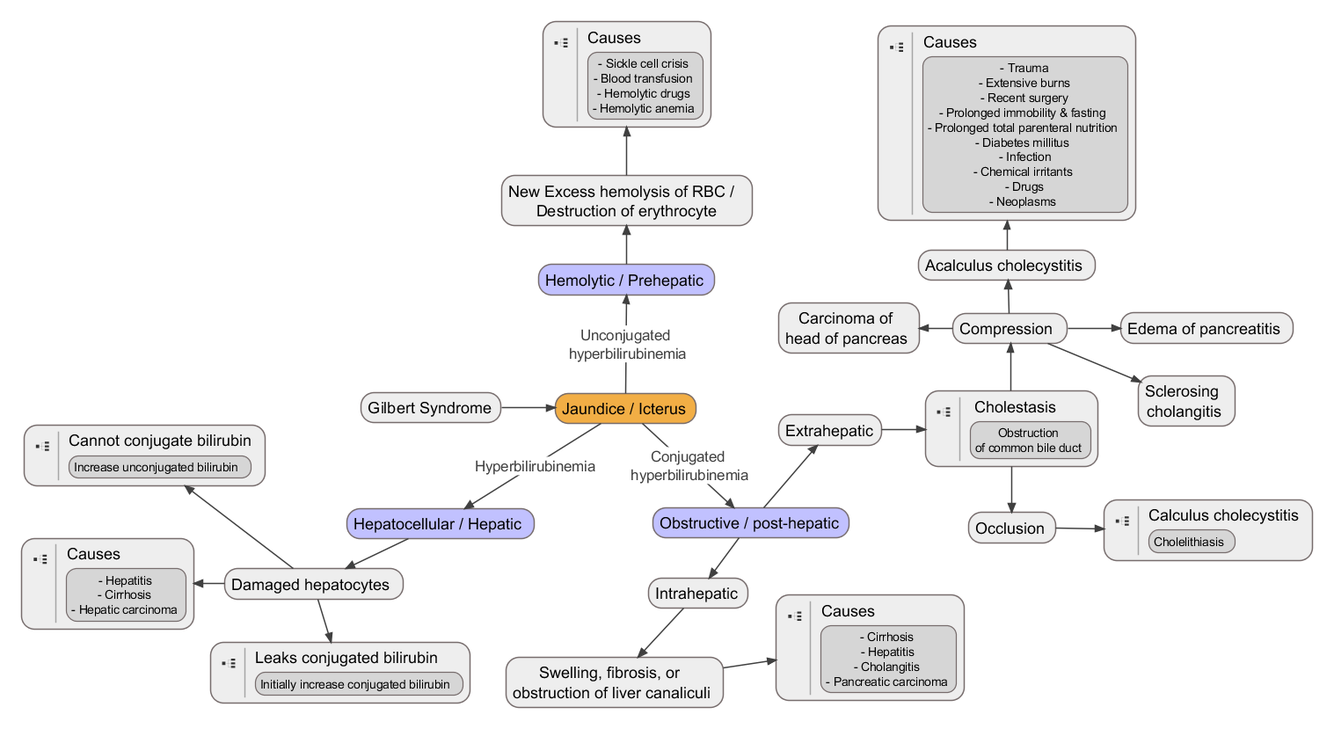
Jaundice: Metabolism of heme, and what are they conjucated with?Classification and differential diagnosis, symptoms, treatment
Neonatal jaundice
Jaundice, also known as icterus, is a yellowish or greenish pigmentation of the skin and whites of the eyes due to high bilirubin levels. The feces may be pale and the urine dark.
Jaundice in babies occurs in over half in the first week following birth and in most is not a problem. If bilirubin levels in babies are very high for too long a type of brain damage, known as kernicterus, may occur.
High bilirubin is divided into two types: unconjugated (indirect) and conjugated (direct). Conjugated bilirubin can be confirmed by finding bilirubin in the urine. High unconjugated bilirubin may be due to excess red blood cell breakdown (e.g. hemolytic anemia), large bruises, genetic conditions such as Gilbert’s syndrome, no eating for a prolonged period of time, newborn jaundice, or thyroid problems. High conjugated bilirubin may be due to liver diseases such as cirrhosis or hepatitis, infections, medications, or blockage of the bile duct.
The main symptom of jaundice is a yellowish discoloration of the white area of the eye and the skin. Slight increases in serum bilirubin are best detected by examining the sclerae, which have a particular affinity for bilirubin due to their high elastin content. Newborns are especially vulnerable to hyperbilirubinemia-induced neurological damage and therefore must be carefully monitored for alterations in their serum bilirubin levels.
Classification
- Prehepatic (unconjugated): Certain genetic diseases, such as sickle cell anemia, spherocytosis, thalassemia, pyruvate kinase deficiency, and glucose 6-phosphate dehydrogenase deficiency can lead to increased red cell lysis and therefore hemolytic jaundice
- Hepatic unconjugates:
- Hepatic conjugated:
- Posthepatic (obstructive): most common causes are gallstones in the common bile duct, and pancreatic cancer in the head of the pancreas
In jaundice secondary to hemolysis, the increased production of bilirubin leads to the increased production of urine-urobilinogen. Bilirubin is not usually found in the urine because unconjugated bilirubin is not water-soluble
The hemoglobin is phagocytosed by macrophages, and split into its heme and globin portions. The globin portion, a protein, is degraded into amino acids and plays no role in jaundice. Two reactions then take place with the heme molecule. The first oxidation reaction is catalyzed by the microsomal enzyme heme oxygenase and results in biliverdin (green color pigment), iron and carbon monoxide. The next step is the reduction of biliverdin to a yellow color tetrapyrol pigment called bilirubin by cytosolic enzyme biliverdin reductase. This bilirubin is “unconjugated,” “free” or “indirect” bilirubin.
The unconjugated bilirubin then travels to the liver through the bloodstream. Because this bilirubin is not soluble, however, it is transported through the blood bound to serum albumin. Once it arrives at the liver, it is conjugated with glucuronic acid (to form bilirubin diglucuronide, or just “conjugated bilirubin”) to become more water-soluble. The reaction is catalyzed by the enzyme UDP-glucuronyl transferase.
This conjugated bilirubin is excreted from the liver into the biliary and cystic ducts as part of bile. Intestinal bacteria convert the bilirubin into urobilinogen. From here urobilinogen can take two pathways. It can either be further converted into stercobilinogen, which is then oxidized to stercobilin and passed out in the feces, or it can be reabsorbed by the intestinal cells, transported in the blood to the kidneys, and passed out in the urine as the oxidised product urobilin. Stercobilin and urobilin are the products responsible for the coloration of feces and urine, respectively
Neonatal jaundice is usually harmless: this condition is often seen in infants around the second day after birth, lasting until day 8 in normal births, or to around day 14 in premature births. Typical causes for neonatal jaundice include normal physiologic jaundice, jaundice due to formula supplementation,[21] and hemolytic disorders that include hereditary spherocytosis, glucose-6-phosphate dehydrogenase deficiency, pyruvate kinase deficiency, ABO/Rh blood type autoantibodies, or infantile pyknocytosis. Serum bilirubin normally drops to a low level without any intervention required. In cases where bilirubin rises higher, a brain-damaging condition known as kernicterus can occur, leading to significant disability.[22] This condition has been rising in recent years due to less time spent outdoors. A Bili light is often the tool used for early treatment, which often consists of exposing the baby to intensive phototherapy. Sunbathing is effective treatment,[23][24] and has the advantage of ultra-violet-B, which promotes Vitamin D production.[25] Bilirubin count is lowered through bowel movements and urination, so frequent and effective feedings are especially important.[26]