HYPOMELANOSIS OF ITO
Genetics: unknown
Inheritance: de novo (typically)
Clinical Features: unilateral or bilateral macular hypo- or hyperpigmented whorls, streaks and patches (sometimes following lines of Blaschko), hair and tooth anomalies are common, ocular abnormalities (strabismus, nystagmus), musculoskeletal system (growth asymmetry, syndactyly, polydactyly, clinodactyly, scoliosis), CNS abnormalities (microcephaly, seizures, ID), cardiac defects
Investigations: R/O Incontinentia pigmenti, which is genetically inherited and requires genetic counselling in future
Management: symptomatic
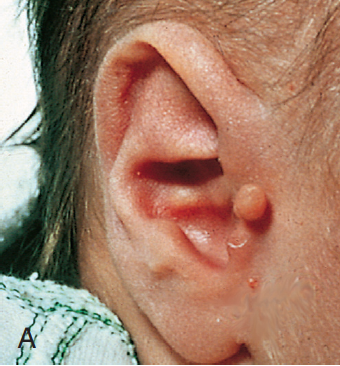
Preauricular skin tag
Associated with:
- Goldenhar syndrome
- Treacher-Collins syndrome
- Wolf-Hirschhorn syndrome
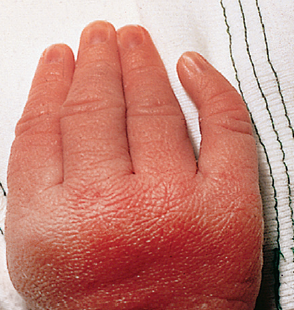
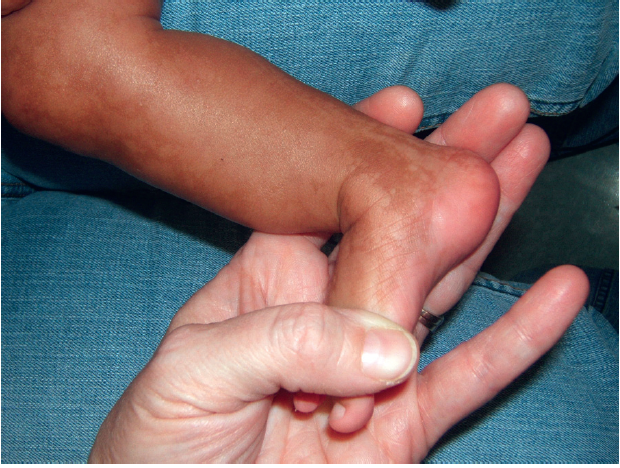
Clinodactyly of the 5th finger
Associated with:
- Trisomy 21
- Normal familial variant
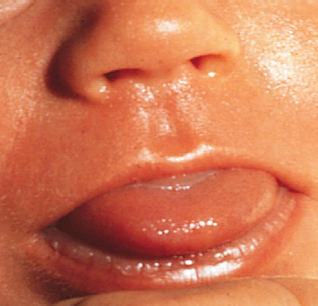
Macroglossia
Associated with:
- Beckwith Wiedemann syndrome
- Mucopolysaccharidosis
- Neurofibromatosis
- Glycogen storage disease - type 2
- Klippel-Trenaunay-Weber syndrome
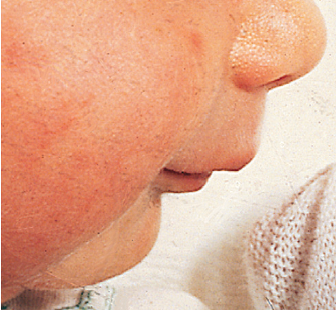
Microretrognathia
Associated with:
- Pierre Robin sequence
- Treacher-collins syndrome
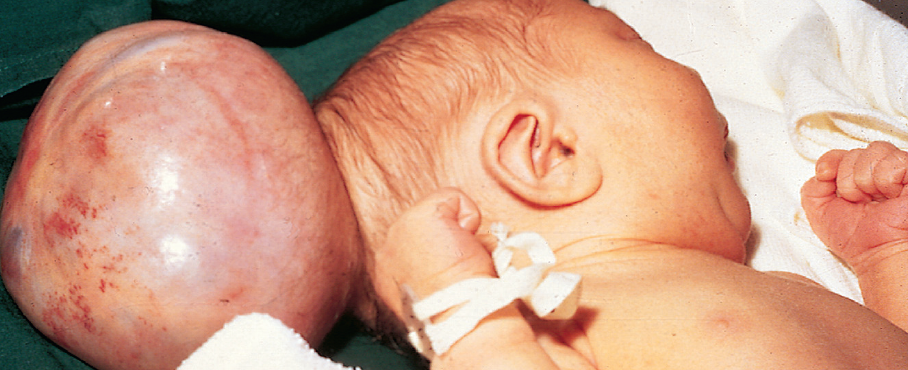
Encephalocele
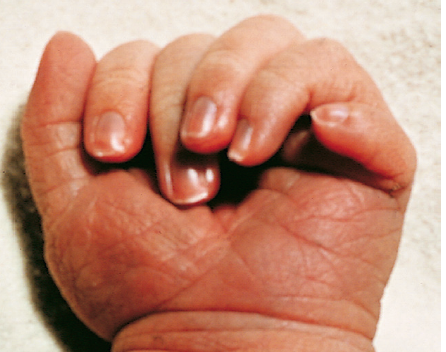
Postaxial polydactyly
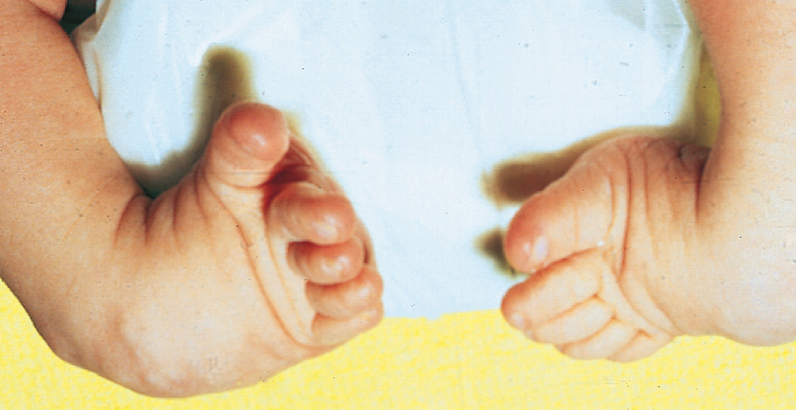
Bilateral clubfoot
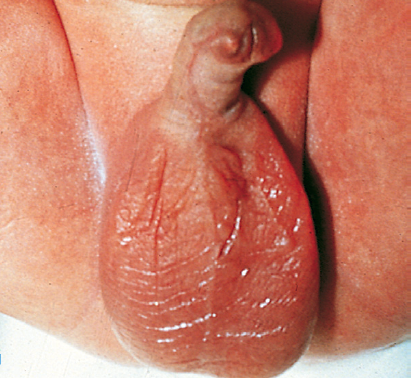
Hypospadias
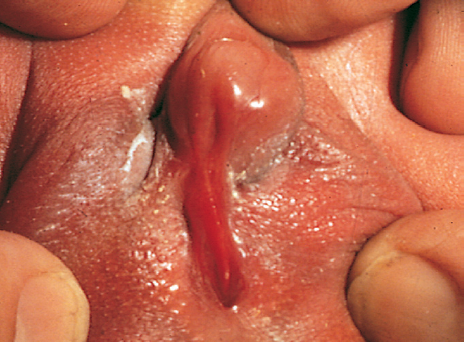
Fused labia with clitoromegaly
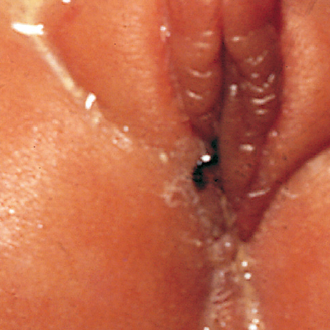
Imperforate anus
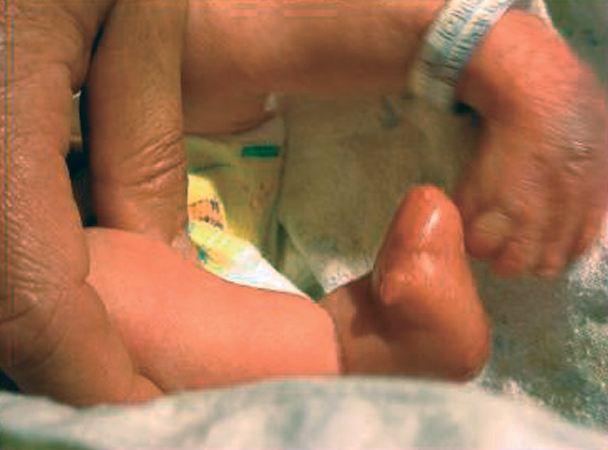
Amniotic band syndrome
Happy child, wide open mouth, seizures

ANGELMAN SYNDROME
Genetics: UBE3A gene deletion/mutation on Ch 15
Imprinting disorder (lack of maternal contribution/uniparental disomy of paternal gene)
Clinical Features: Microcephaly, hand flapping, ADHD, atypical laughing/smiling, Seizures, Hypopigmentation (skin/eyes), smooth palms, increased sensitivity to heat, prominent mandible, wide mouth, protruding tongue, arm tremors, jerky movements
Investigations:
- Methylation testing of Chromosome 15
Monitoring
- Hyperactivity and poor sleep improves over time
- Seizures escalate around time of puberty (especially in girls)
- avoid carbamazepine, vigabatrin and tiagabine
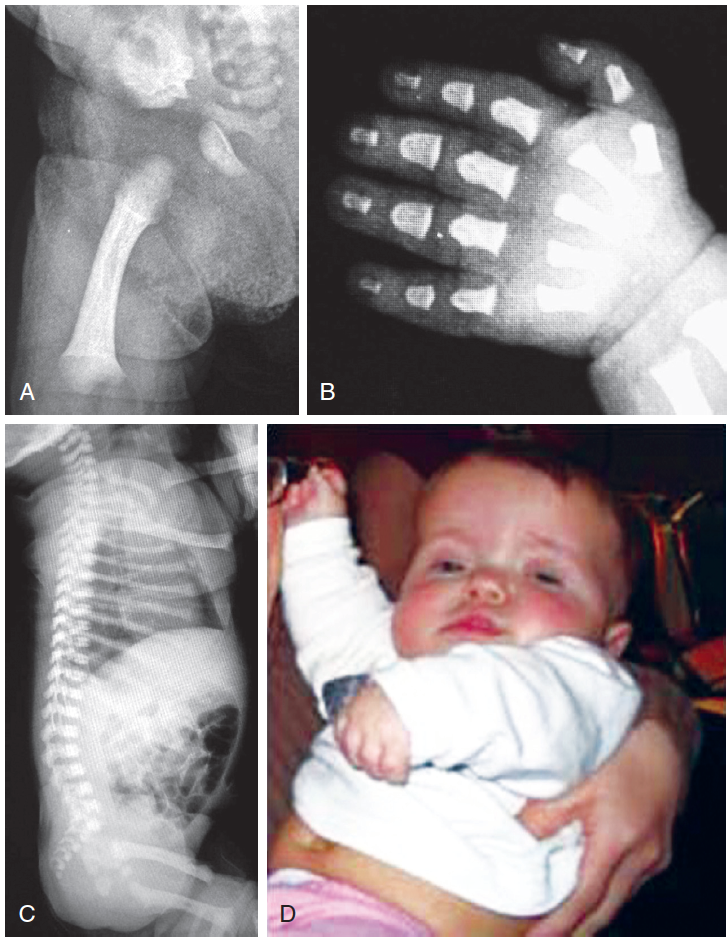
ACHONDROPLASIA
Gene: FGFR3 gene, codon 380
Inheritance: behaves Autosomal Dominant
Clinical features: Frontal bossing, depressed nasal bridge, sausage fingers, disproportionate size (short extremities, large head), small chest, protruding belly, trident hand
Associations:
- Hydrocephalus (d/t foramen magnum stenosis)
- Middle ear dysfunction (40% hearing loss, frequent AOM)
- Delayed motor milestones (often not walking until 18-24mo)
- Obstructive sleep apnea
- Delayed speech + articulation difficulties
- Dental crowding
- Bowing of legs (may need surgical correction)
- Obesity
Work-up
- Skeletal radiographs (short vetebral pedicles, large calvarial bones)
- Genetic testing
Long-term
- Monitor for developmental delay, scoliosis, arthritis, hydrocephalus
- Referral to ENT, dentistriy
*
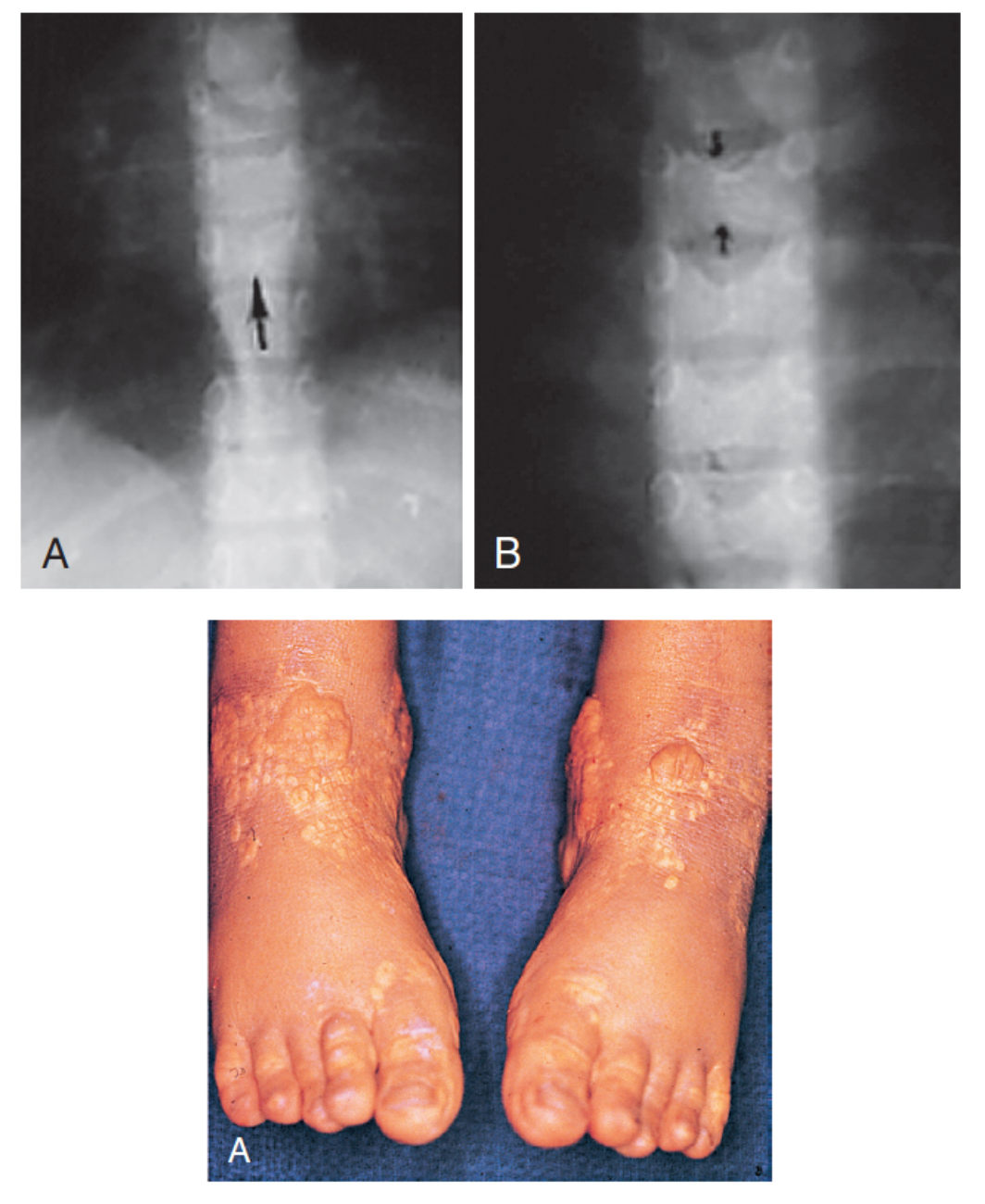
ALAGILLE SYNDROME
Genetics: JAG1, NOTCH2 mutations
Inheritance: Autosomal Dominant
Clinical Features: Butterfly vertebrae (clefting, failure of fusion), Posterior embryotoxon, Conjugated hyperbilirubin due to bile duct paucity, Peripheral pulmonary artery steonosis, renal disease, pancreatic insufficiency, growth delay, ID/GDD
Investigations:
- Radiographs: XR spine
- Ultrasound of gallbladder/HIDA
- Echocardiogram
- Genetics - sequence analysis of JAG1/NOTCH2
Monitoring
- Multidisciplinary (Genetics, GI, Nutrition, Nephro, Ophtho, Cardio)
- Ursodiol for cholestasis
- Liver transplant for ESRD
- Fat soluble vitamin supplementation
- Avoid contact sports and alcohol
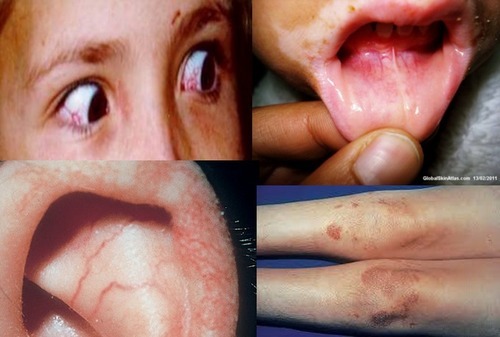
ATAXIA TELANGIECTASIA
Inheritance: Autosomal Recessive
Complex immunodeficiency disorder with DNA repair defect
Clinical Features: Initially normal, develop ataxia ~2-3yo [usually first 6y of life] (wheelchair bound by 15yo), oculomotor apraxia (cannot make fast eye movements), Telangiectasia (last to appear), Immunodeficiency (decreased Ig, T-cell dysfunction), Malignancy (leukemia, lymphoma), recurrent sinus/pulmonary infections can lead to bronchiectasis
Investigations:
- Elevated AFP
- Low serum IgA
- Genetic testing
Monitoring:
- Supportive
- Death in 20s
Macroglossia, hemihypertrophy, omphalocele
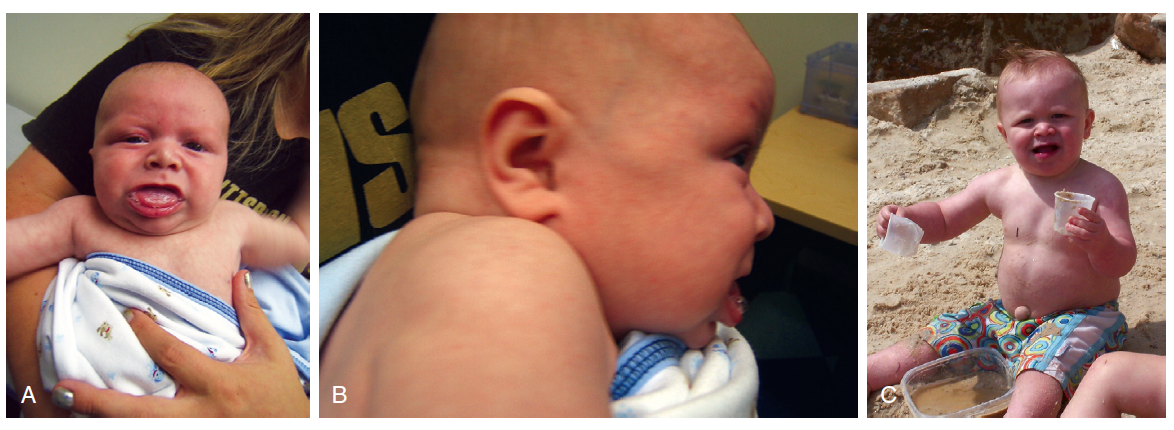
BECKWITH WIEDEMANN SYNDROME
Inheritance: Imprinting disorder (Ch11p15); Autosomal dominant
Higher risk in IVF pregnancies
Clinical Features: Polyhydramnios, LGA baby, Macroglossia, Abdominal wall defects (omphalocele), pre-auricular ear creases/pits, renal abnormalities, hemi-hypertrophy, hyperplasia of organs, renal abnormalities, neoplasms (Wilms, adrenal carcinoma, hepatoblastoma)
Investigations: Chromosomal microarray
Monitoring:
- Hypoglycemia (infants)
- Abdo US q3mo until 8yo
- Serum AFP q3mo until 4yo
- CXR periodic (thoracic neuroblastoma)
- Renal US annually (8-16y)
- Ortho (hemihypertrophy)

CHARGE SYNDROME
Genetic: CHD7 mutations
Inheritance: Autosomal Dominant
Clinical Features:
- Coloboma
- Heart defect (conotruncal, AV canal, aortic arch)
- Atresia choanae (TEF, cleft lip and palate)
- Retardation of growth (short +/- GH deficiency)
- GU anomalies (single kidney, hydronephrosis, renal hypoplasia, micropenis, hypoplastic labia, cryptorchidism)
- Ear anomalies (question mark ear)
Can have facial asymmetry due to CNVII palsy, square face with flat midface, broad nose, swallowing difficulties due to CN abnormalities.
Investigations: Genetic testing, echocardiogram, abdominal U/S
Management:
- ENT, Ophtho, Nephro/Urology, Cardiology referral
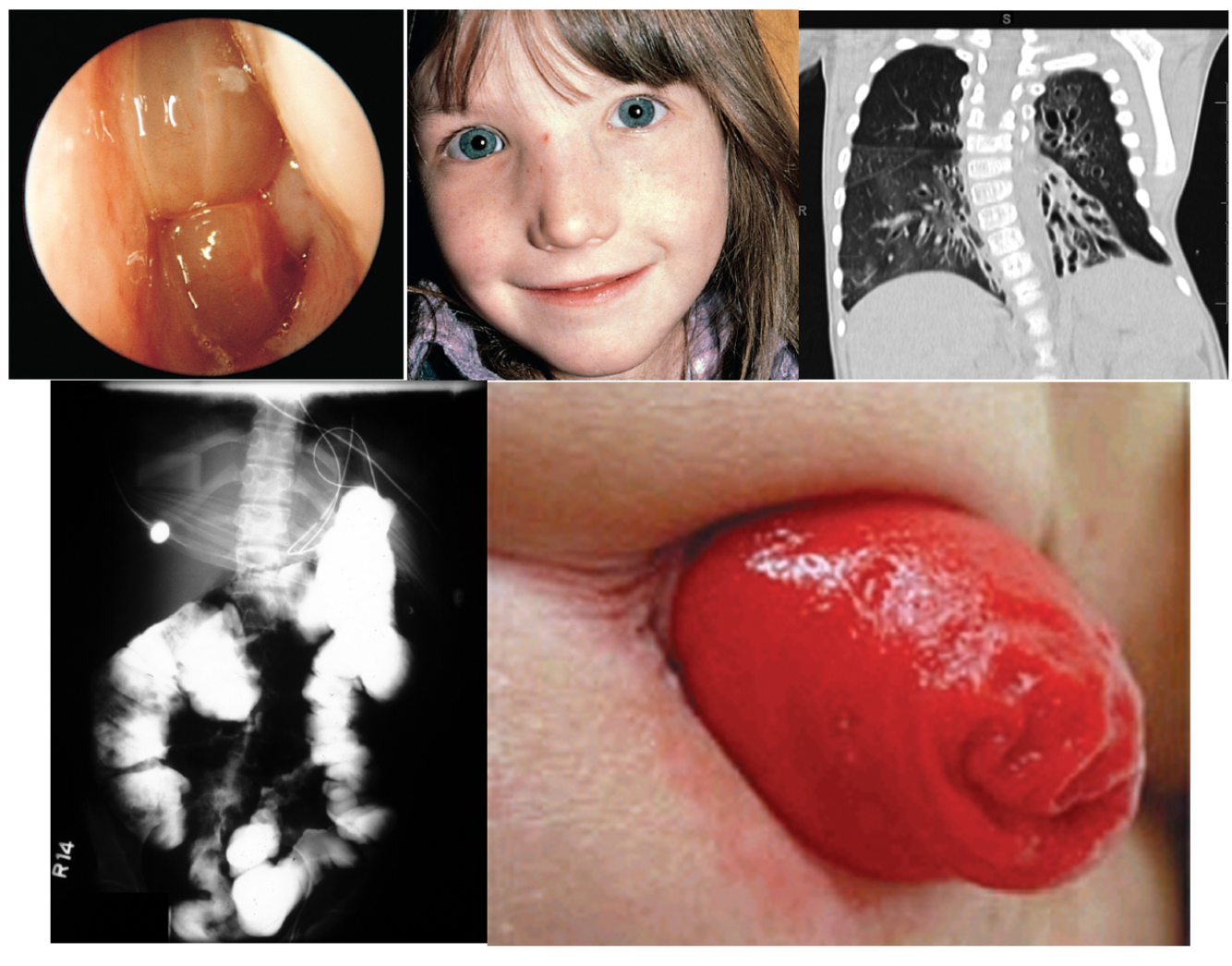
CYSTIC FIBROSIS
Genetics: gene that codes for the CFTR protein (majority are ΔF508)
Inheritance: Autosomal Recessive
Mechanism: CFTR dysfunction = ↓Cl secretion and ↑Na absorption, leading to dehydrated/viscous mucus
Clinical Features (I’m CF Pancreas)
- Infertility
- Meconium ileus
- Cough
- Failure to thrive
- Pancreatic insufficiency
- Asthma (refractory)
- Nasal polyps
- Clubbing
- Rectal prolapse
- Electrolyte abnormalities (metabolic alkalosis, ↓Na, ↓Cl, ↓K)
- Atypical organisms from sputum
- Sludge (cholelithiasis/cystitis, pancreatitis, sinusitis)
Respiratory: bronchiectasis, pneumothorax, respiratory failure
Gastrointestional: DIOS, intussusception, biliary cirrhosis, hepatic steatosis, GERD, inguinal hernia, steatorrhea, fat-soluble vitamin deficiency (A, D, E, K)
Delayed puberty, hypertrophic osteoarthropathy/arthritis, amyloidosis, aquagenic palmoplantar keratoderma (skin wrinkling), hypoproteinemia
Diagnosis:
Requires clinical features OR sibling with CF OR positive NBS
AND
Elevated sweat chloride OR abnormal nasal potential difference OR identification of 2 disease-causing CF mutations
Management:
- Suppressive antibiotic therapy
- Mucociliary clearance (chest PT)
- inhaled mucolytics (DNase if >6yo)
- Bronchodilators
- Inhaled 3%NaCl
- Antiinflammatory: NSAIDs and macrolides (3x/wk)
- Nutrition: enzyme replacement, vitamin supplements, high-fat, high protein diet, MCTs added
- Insulin PRN
- Ursodiol to prevent/treat liver disease
- CFTR modulators
- Lung transplant
Characterized by proteinuria, edema, ambiguous genitalia. What to screen for?
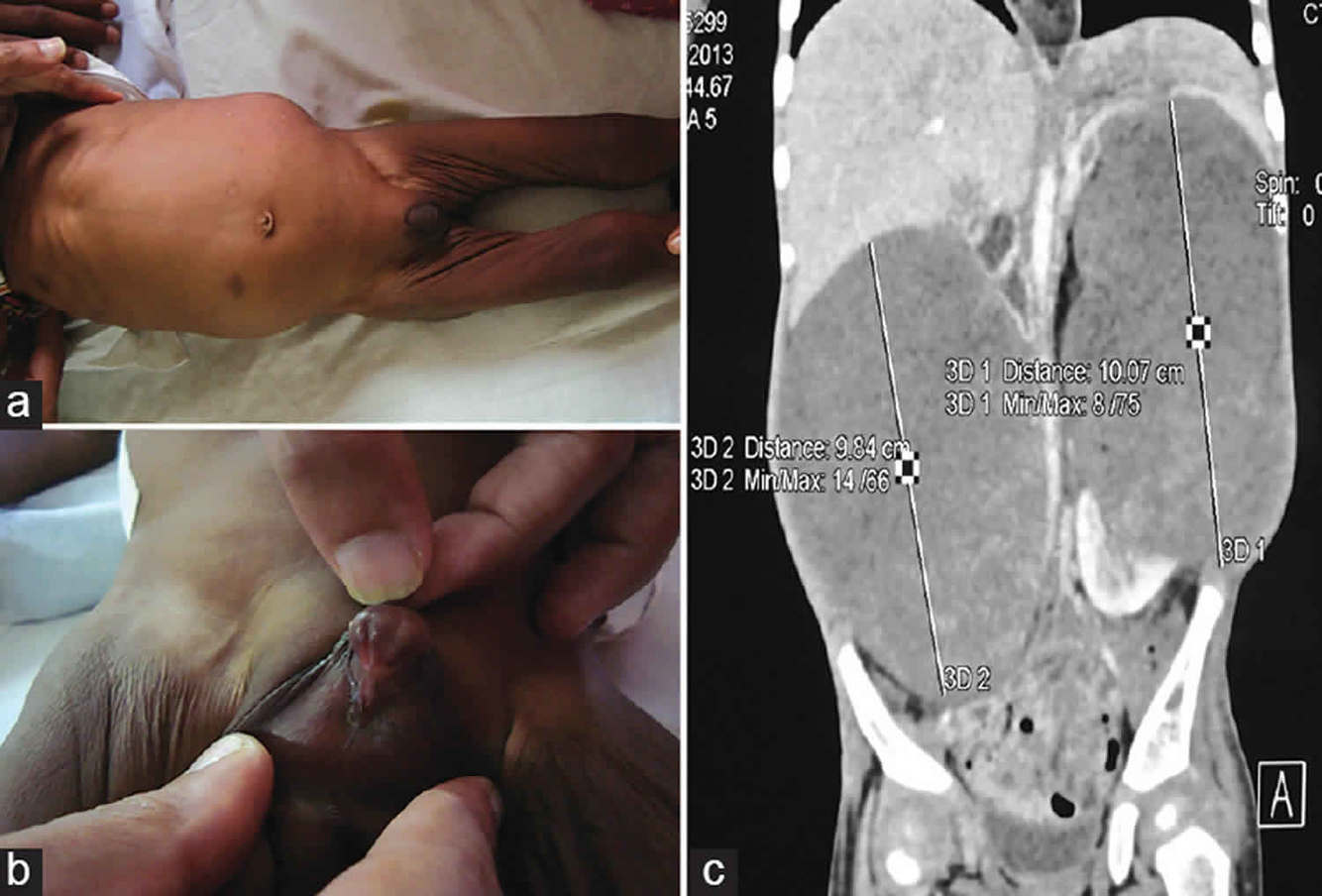
DENYS-DRASH SYNDROME
Genetics: WT1 gene mutation
Clinical Features:
- Nephropathy
- Ambiguous genitalia
- Bilateral Wilms tumours (<2yo)
- Proteinuria in infancy → nephrotic syndrome (edema) → ESRD
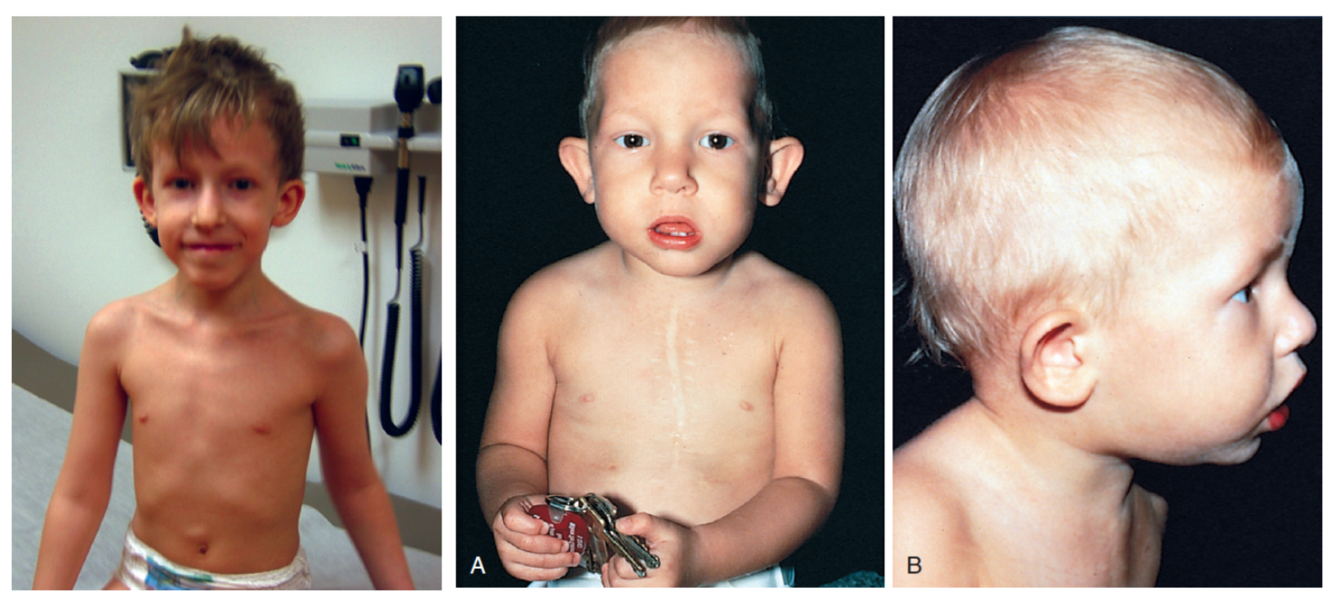
DiGEORGE SYNDROME
Genetics: 22q11.2 microdeletion
Clinical Features:
- Cardiac abnormalities (TOF most commonly)
- Abnormal facies (malar hypoplasia, square face, mild hypertelorism, prominent ears)
- Thymic hypoplasia + immunodeficiency
- Cleft palate + velopharyngeal insufficiency
- Hypocalcemia, hypoPTH
- 22 chromosome
Learning difficulties/ID, psychiatric issues (schizophrenia), hearing loss
Investigations: serum calcium, echocardiogram, chromosomal microarray
Management:
- Referral to Audiology, Cardiology, Ophthalmology, Immunology
- Repeat calcium levels q3-6mo, TSH, PTH
- Immune function testing
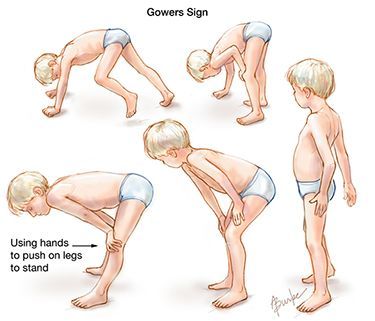
DUCHENNE MUSCULAR DYSTROPHY
Genetics: Dystrophin gene mutation
Inheritance: X-linked recessive
Clinical Features: Presents at 2-3yo; proximal > distal muscle weakness, lower extremities > upper extremities, Gowers sign, Calf pseudohypertrophy, Cardiomyopathy (~15yo), Fractures, Scoliosis, Impaired pulmonary function, Obstructive sleep apnea, decreased gastric motility
Investigations: ↑CK, EMG abnormal, muscle biopsy, genetic testing for dystrophin gene (molecular)
Confined to wheelchair by age 12, death in 20s
Management:
- Multidisciplinary Neuromuscular clinic: Neurology, Rehab, Cardiology, Orthopedics, Respirology, Physiotherapy, Bone health
- Steroids to try and prolong course (↑motor function, ↑pulmonary function, ↓development of cardiomyopathy, ↓scoliosis)
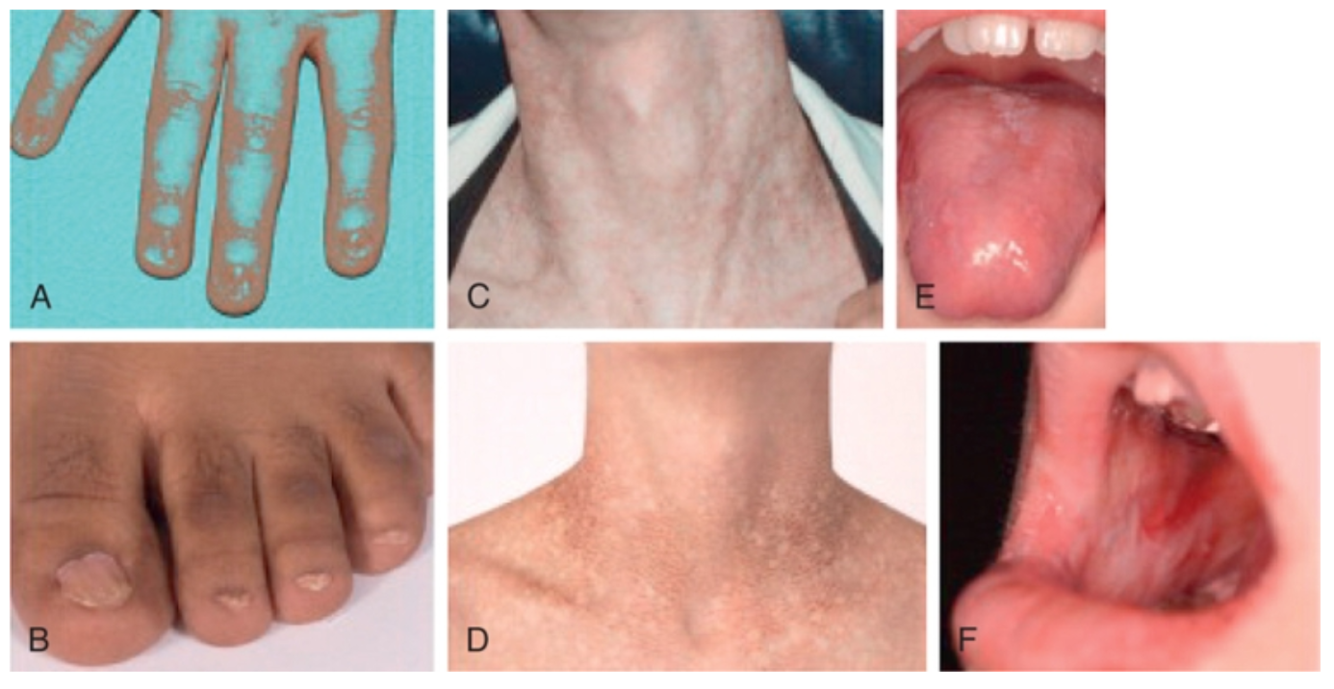
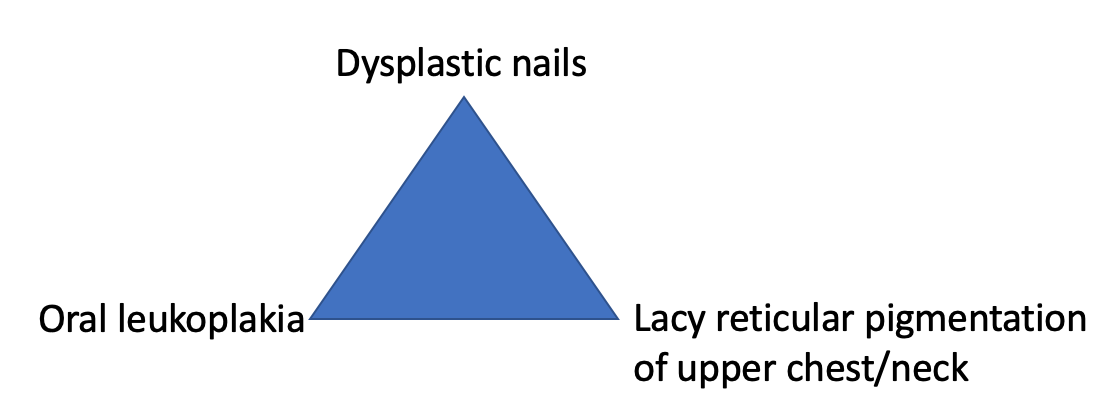
DYSKERATOSIS CONGENITA
Inherited multisystem telomere disorder. (AD and AR)
MAJOR Features:
- Abnormal skin pigmentation
- Nail dystrophy
- Leukoplakia (usually tongue, can involve conjunctiva, anal, urethral or genital mucosa)
- Bone marrow failure
Clinical features: Some genetic types are at risk of pulmonary/hepatic fibrosis. Can have excessive tearing. 25% have LD/ID. Short stature in 15-20%
Investigations: Telomere length study. CBC to evaluate for bone marrow failure.
Management:
- Cancer predisposition (possible): solid tumours, MDS, AML
- Androgen therapy
- Bone marrow transplant
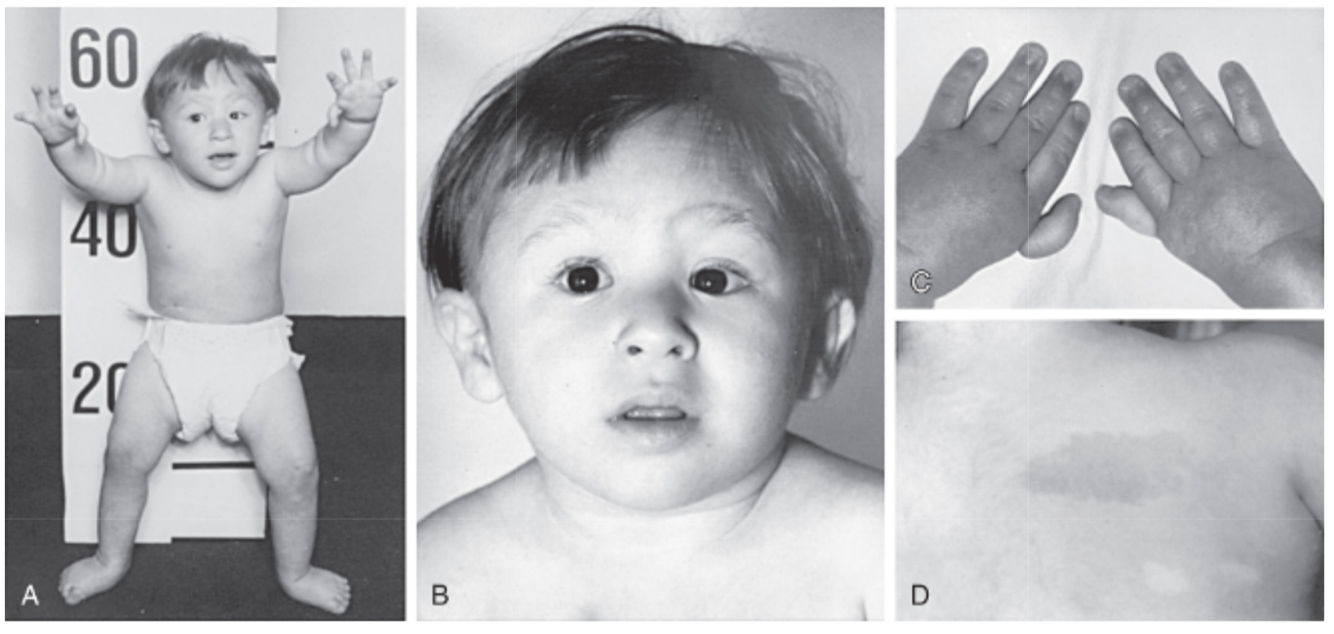
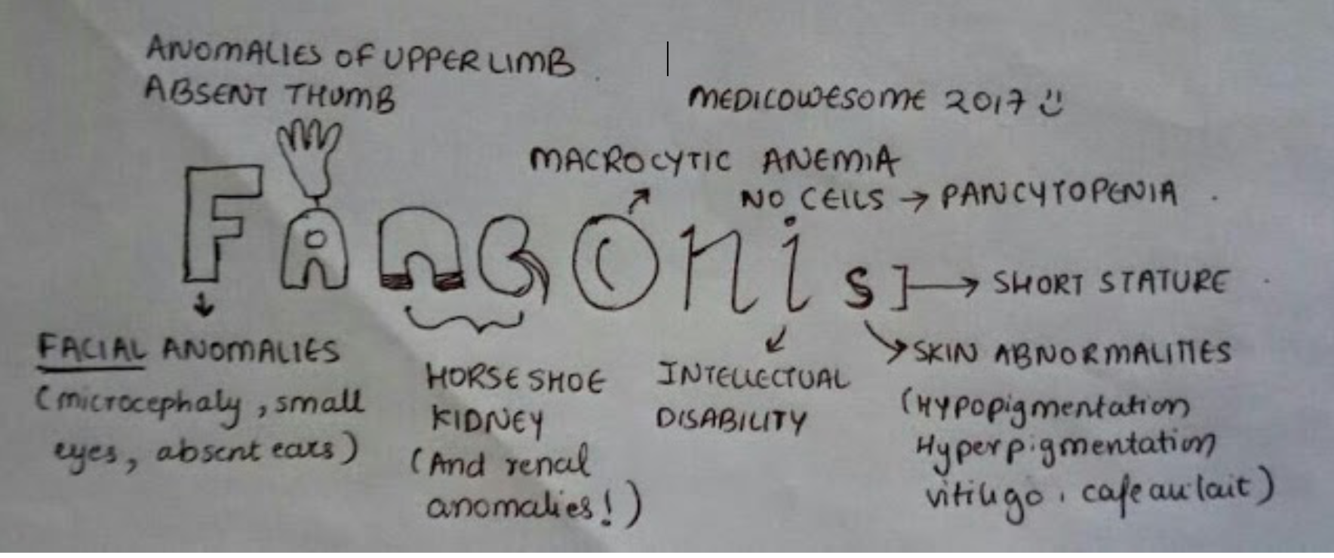
FANCONI ANEMIA
Genetic: FANC genes
Inheritance: X-linked recessive (most common)
Consider on differential for any unexplained cytopenia.
MINIMIZE RADIATION EXPOSURE because of carcinogenic risk
Bone marrow failure appears within 1st decade of life.
(↓platelets, ↑MCV, ↑HgbF appear first → neutropenia → anemia)
Clinical Features:
- Skeletal (absence of radii and/or thumb abnormalities [hypoplastic, supernumerary, bifid or absent], feet or leg anomalies, congenital hip dislocation)
- Skin hyperpigmentation of trunk, neck and skin folds, CALMs, vitiligo (alone or in combo)
- Short stature +/- GH deficiency or hypothyroidism
- Dysmorphic features: microcephaly, epicanthal folds, small eyes, abnormal shzpe, size or positioning
- Males (all infertile): underdeveloped penis, undescended, atrophic or absent testes, hypospadias or phimosis
- Females: reduced fertility, malformations of ovary, uterus and ovary
- 10% ID
- IUGR/LBW
Predisposition to MDS (myelodysplasia), AML and SCC.
Investigations:
- Lymphocyte chromosomal breakage study
- Imaging: U/S abdomen, echocardiogram
- If short stature - work-up for GH deficiency
- Blood work should include: liver, thyroid, metabolic and immune system
Management:
- HSCT - only curative therapy
- Androgen therapy
- Referrals if abnormalities identified
- Multidisciplinary team including a Hematologist
- Mild-moderate CBC AbN + no transfusion = CBC q3mo + annual BMA + BMBx PRN
- Glucose levels q6mo for hyperglycemia
- TSH annually
- Solid tumour screen with physical exam annually















































-
Acute Care270
-
Adolescent/Gyne98
-
Allergy/Immunology70
-
Cardiology136
-
CYPT and Ethics64
-
Dermatology70
-
Endocrinology161
-
ENT/Ophtho/Gen Surg149
-
Gen Paeds0
-
Genetics179
-
GI/Nutrition146
-
Heme/Onc106
-
Infectious Disease271
-
Metabolics27
-
MSK/Ortho/Rheumatology138
-
Nephrology/Urology154
-
Neurology110
-
NICU233
-
Pharm and Critical Appraisal57
-
Psych/Development200
-
Respirology152
-
ADHD13
-
Syndromes102
