What are the differnt types of hemoglobin ? What is heme? How many heme are there in one chain and how many per molecule of Hb?
- Hemoglobin (Hb) is a tetrameric protein made up of two types of polypeptide chains (Globin chains)
- Adult Hb (HbA)-Main Hb: 2 α- chains and 2 β-chains.
- Fetal Hb (HbF): about 1-2%, - 2α and 2γ chains
- HbA2 : about 2% made up of 2α and 2δ chains
- Heme (Fe++ containing protoporphyrin) is the non protein prosthetic group
- Each chain contains one Heme (Totally 4 Heme per molecule of Hb)
Howm any amino acids are in each chain ? How many a-helical segments are there in each chain ? How are polar and non-polar groups distributed in hemoglobin? What are the heme groups surrounded by? What are the role of the proximal and distal histidines?
•α-chain has 141 & β, chain has 146 amino acids.
•Both the chains contain extensive α- helix structures
•7 α- helical segments (A to G) in α- chain & 8 α- helical segments (A to H) in β-chain
•Interior of each globin chain has non polar amino acids
•Polar a.a. residues (including basic and acidic) are on the surface.
•Heme group occupies a crevice surrounded by non polar residues and 2 histidines; E-7 & F-8.
•F-8 His (proximal histidine) is closer to heme and binds Fe2+ and E-7 His (distal histidine) stabilizes O2 bound to Fe2+
Hemoglobin is a tetramer made of two dimers. What are the dimers the polypeptides are mainly held together by? What are the dimers held by? What are the two forms of hemoglobin? Which form has high and low affinities for hemoglobin?
- Hemoglobin is a tetramer, made up of dimers (αβ)1 and (αβ)2.
- In each of dimer the two polypeptides are held together mainly by hydrophobic (non polar) & partly by ionic and hydrogen bonds
- Two dimers ((αβ)1 and (αβ)2) are held together by ionic & polar bonds with freedom of movement between them
•Relative positions of dimers makes Hb to occur in two forms; T-form (taut or tense) and R-form (relaxed)
•In T-form the network of ionic and H-bonds are stronger and movement is limited. This form has low affinity to oxygen – oxygen unloading -forms deoxy-Hb
•R-form has high affinity to oxygen, more freedom of movement for polypeptide chains – oxygen loading – forms oxy-Hb
What is myoglobin? Where is it mainly present? What type of structures does it contain? How is myoglobin different form hemoglobin? What is the shape of myoglobin dissociation curve? Since the curve for Hb is sigmoidal, what does this suggest?
- Myoglobin is a heme protein structurally similar to Hb but consists of single globin chain and one heme group. It is present in heart and skeletal muscles.
- Has extensive α-helical structure (similar to Hb) . Contains 8 α-helices.
- Myoglobin (Mb) binds one molecule of O2 unlike Hb (binds 4O2, one each at 4 heme groups)
- Oxygen dissociation curve for Mb has hyperbolic shape.
- The curve for Hb is sigmoidal suggesting cooperative binding of O2. Binding of O2 on one heme increases affinity of other heme groups for oxygen.
What are the factors that influence oxygen loading and unloading (4)? These effects are collectively also called?What is cooperativity? What is Bohr’s effect ? When there is high pH and lower partial pressure of CO2, what happens to the affinity for oxygen towards Hb?
- Unloading and loading of oxygen to Hb is affected by partial pressure of O2, pH, partial pressure of CO2 and the presence of 2,3-bisphospho-glycerate (2,3 BPG)
- These effects are collectively called allosteric effects.
- Binding of O2 to one of the tetramer enhances the affinity of heme for the binding of next O2. This is called cooperativity (due to heme- heme interaction)
- The influence of low pH and increased partial pressure of CO2 in unloading of oxygen from oxyHb is called as Bohr effect
- Conversely, higher pH and lower partial pressure of CO2 results in greateraffinity for oxygen towards Hb
What is the importance of 2,3,BPG? What conformation does it favor? What is the 2,3BPG binding site created by? How is it formed? When is the concentration of 2,3, BPG increased(3)?
- It is an important regulator of O2 binding to Hb.
- It decreases oxygen affinity to Hb. Favors T conformation
- 2,3 BPG binding site is created by two β- globin chains of Hb
- 2,3 BPG is formed from an intermediate of glycolysis (1,3 BPG)
- The conc. of 2,3 BPG is increased in response to hypoxia as seen in
- — Pulmonary obstructive disorders
- — High altitude
- Increase is also seen in chronic anemia (due increased demand of O2 by tissues)
- Improperly preserved blood (in blood banks) may be depleted of 2,3 BPG
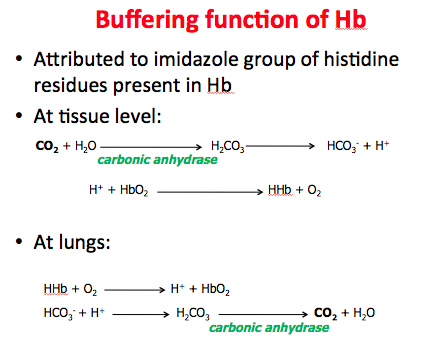
Explain the buffering function of hemoglobin!
What are the other aspects of hemoglobin?
- Other aspects of Hb:
- Hb can reversibly bind nitric oxide (NO), a potent vasodilator
- Hb is involved in the transportation of carbon dioxide as carbamino Hb
- Carboxy Hb (Hb with CO) – Carbon monoxide (CO) has over 220 times higher affinity to Hb than O2.
How does the fetal hemoglobin differ from adult? (4)
- Major Hb of fetus and newborn
- 2 α 2 γ chains
- Replaced by HbA after birth
- Has high affinity to oxygen
- Weakly binds to 2,3 BPG (due to the absence of β chain)
- Higher affinity to oxygen facilitates HbF to transfer oxygen from maternal circulation to the fetus across the placenta
What is glycated hemoglobin represented as? What happens in glycated hemoglobin ?
Describe the normal range , the pre-diabetic and in diabetes mellitus
- Glycated Hemoglobin:
- It is represented as HbA1c
- Free glucose residues bind to N-terminal a.a. residues of globin chains
- The process is called as non- enzymatic glycosylation
- **In normals 4 – 5.6 % of Hb exists as Hb A1c,
- Pre-diabetic individuals: 5.7 – 6.5%
- Diabetes mellitus: more than 6.5%**
- The measurement of HbA1c indicates average glucose conc. in plasma over a period of approx. past 2 months.
What are hemoglobinopathies? Distinguish the classification of hemoglobinopathies(2)
**Disorders of human hemoglobin **
•Hemoglobinopathies represent a group of most common single gene diseases
•Molecular and biochemical aspects of hemoglobinopathies are well understood
•Classification of Hemoglobinopathies
–Structural variants - Having altered globin chain
–Thalassemias - Decreased synthesis of either α or β globin chains, resulting in imbalanced amount of α and β globin chains
What does Hemoglobin S cause? What is the mode of inheritance? Explain in terms of the amino acid that has been changed in this disease. What are some changes that occur in the RBC? Which group is mostly affected?
- Causes sickle cell disease (sickle cell anemia)
- Geographic distribution – Mainly in malaria endemic areas
- Autosomal recessive mode of inheritance
- Single nucleotide substitution in β globin gene (GAG à GTG) à Replacement of Glutamate by Valine at 6th position in β globin chain
- Loss of a negative charge
- Decreased solubility of the HbS under low pO2
- HbS polymerizes in rod shaped form
- Distorts shape of RBC to sickle shape under low pO2
- RBC unable to squeeze through capillaries
- Homozygotes are severely affected: vascular occlusion, hypoxia, severe pain, hemolytic anemia, Splenic sequestration
Presene of what is advantagous to sickle cell anemia? What are some of the sickle cell heterozygote traits see? How is it seen in? What type of resistance do heterozygous state of Hb S confer?
•Presence of HbF is advantageous in sickle cell anemia- prevents polymerization of Hbs & thereby prevents sickling
•Sicke cell Heterozygotes (Sickle cell traits)
- Are clinically normal, in general - RBC may become sickle shaped in vitro at very low oxygen tension - Risk of splenic infarction while flying at high altitude with reduced cabin pressure of aircraft
- Seen in about 8% among African Americans
- Up to 25% among the population of central Africa
- Heterozygous state of Hb S confers malaria resistance to the individuals
Hemoglobin C:
What is the mode of inheritance?
Where does the mutation occur?
What is its solubility in terms of Hb A?
What are the characteristics of Hb C?
What other type exist? And how is it different from hemoglobin c?
- Also an example for novel property mutation
- Mode of inheritance – autosomal recessive
- Mutation in β globin, Glu à Lys at 6th position
- Hb C is less soluble than Hb A
- Crystallizes in RBC
- Cause mild hemolysis in capillaries
- More cases among west Africans
- Compound heterozygotes (SC disease) also exist
- SC diseases (Hb SC) show milder hemolytic anemia than HbS
- Hb SC may rarely cause vascular occlusion mainly in retina
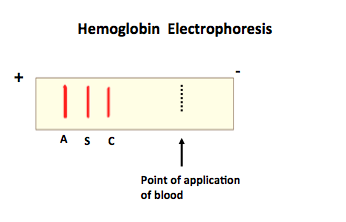
How is the hemoglobin electrophoresis when HbS, HbC, and HbA are compared?
What is methemoglobinemia?
What does it result in ? In normal humans how is methemoglobin converted back into normal Hb( enzyme)?
What may be the cause of acquired methemoglobinemia?
What is the treatment for methemoglobinemia?
What happens in the genetic type of methemoglobinemia?
How is the homozygous differ from the heterozygous?
What is an example of Hb M?
What type of mutation is it and what amino acids are involved?
- Methemoglobin (MetHb)is the one where Heme iron exists in oxidized ferric form (Fe3+)
- Methemoglobin cannot bind O2
- Methemoglobinemia results in cyanosis (bluish)
- Brown colored blood in severe cases
- A small amount of Methemoglobin formed in normal individuals is converted back to normal Hb by NADH – metHb reductase. Deficiency of this enzyme may cause methemoglobinemia
- Acquired methemoglobinemia – may be due to administration of nitrate drugs (treatment of Angina)
- Methemoglobinemia may be treated with methylene blue (MetHb is re-oxidized back to normal Hb)
- Genetic type of Methemoglobinemia(Hb M)
- (due to genetic defect) heme crevice is affected in such away that heme iron is kept in ferric state
- Homozygous state is presumed to be lethal
- Hb Hyde Park(an example for Hb M)
- Point mutation proximal Histidine is replaced by Tyrosine at 92 position in β chain
What are thalassemia?
What are the two types?
What type of anemia does it manifest as?
What do heterozygous individuals acquire form it?
Where is it most commonly seen?
Synthesis of what is defective?
What happens once there is a defect in the production of hemoglobin?
- Represent heterogeneous group of disorders affecting Hemoglobin synthesis
- Collectively most common human single gene disorders
- First discovered in persons of Mediterranean origin
- Two types - α & β thalassemia
- Major manifestations – Hypochromic microcytic anemia
- Carrier (Heterozygous) individuals acquire protective advantage against malaria
- Prevalence – Mainly in Mediterranean, Middle East, Parts of Africa and Asia
- Synthesis of either the α or the β chain is defective (Decreased rate of synthesis or occasionally not synthesized at all)
- Imbalance in the ratio between α & β chains
- Excess normal chains eventually get precipitated within the cell
- Defect in production of hemoglobin
- Damage to cell membrane
- Premature red cell destruction
Describe a-thalassemia.
What two types of disease state does it constitute?
Explain when there is a mutation in one copy, two copies, 3 copies and 4.
What is a-thalassemia mostly caused by?
- α-globin chain production is either decreased or absent
- Constitute both intrauterine and postnatal disease, since the formation of both fetal and adult hemoglobins are affected
- In normal : 4 copies of gene
- Defect (Mutation) in one copy – silent carriers
- Defect in 2 copies - α –Thalassemias trait with mild symptoms
- Defect in 3 copies – HbH disease with variable severity HbH is precipitated – forms Heinz bodies - hemolysis
- Defect in all 4 copies – Hb Bart disease with hydrops fetalis – cause mostly fetal death
- In the absence (deficiency) of α-globin unusual chain combinations take place
- Hb Bart’s – γ4 Hb H - β4
- Both are ineffective as oxygen carriers, however
- α- thalassemia is mostly caused due to deletions in α-globin genes
What is b-thalassemia?
What happens due to this?
What type of anemia can this lead to?
When are the clinical features seen?
Levels of what are elevated in a ppt with b-thalassemia?
How is b-thalassemia differ from a-thalassemia ?
What is affected at the cell’s level?
- Genetic disorders of β-globin production
- Decreased ( or stoppage) β-globin production
- Excessive α –globins precipitate in RBC causing RBC destruction
- Hypochromic , microcytic anemia
- Clinical features seen only a few months after birth because β-globin is important only in postnatal period (to form Hb A)
- Elevation of Hb A2 is unique because δ (delta) globin production is not affected
- Hb F is also increased (because of selective survival)
- β thalassemia is usually caused by point mutations rather than deletion (unlike α thalassemia)
- Mainly affects RNA splicing, thereby decrease the availability of functional β-globin mRNA needed for the synthesis of β-globin chain
Compare B-thalassemia major and b-thalassemia minor
•β -Thalassemia major:
•The individuals who are homozygous to β thalassemia (both the genes are defective)
•Exhibit severe anemia and require life long medical attention
•β0 thalassemia – No Hb A present
•β+ thalassemia – Hb A is detectable
•β -Thalassemia minor:
•Carriers of one β thalassemic gene
•Have hypochromic, microcytic RBC with slight anemia
•β thalassemia minor may be initially misdiagnosed as iron deficiency anemia
-
Metabolism of glycogen (glycogenesis & glycogenolysis)35
-
Basic Aspects of Amino acids20
-
General structure of Proteins25
-
Hemoglobin20
-
Enzymes 128
-
Enzymes 232
-
Carbohydrates- digestion and absorption12
-
Carbohydrates- glycolysis (Part 1)16
-
Carbohydrates- glycolysis (Part 2)11
-
Carbohydrates –pyruvate oxidation & citric acid cycle (TCA cycle)21
-
GLUCONEOGENESIS18
-
HMP Shunt20
-
Metabolism of fructose, galactose & lactose15
-
Glycoaminoglycans25
-
collagen elastin and a1-antitrypsin12
