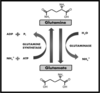
Common GBA mutations
AJew: N409S (formerly N370S)
Europe: L483P (formerly L444P)
- Original nomenclature not include first 39AAs
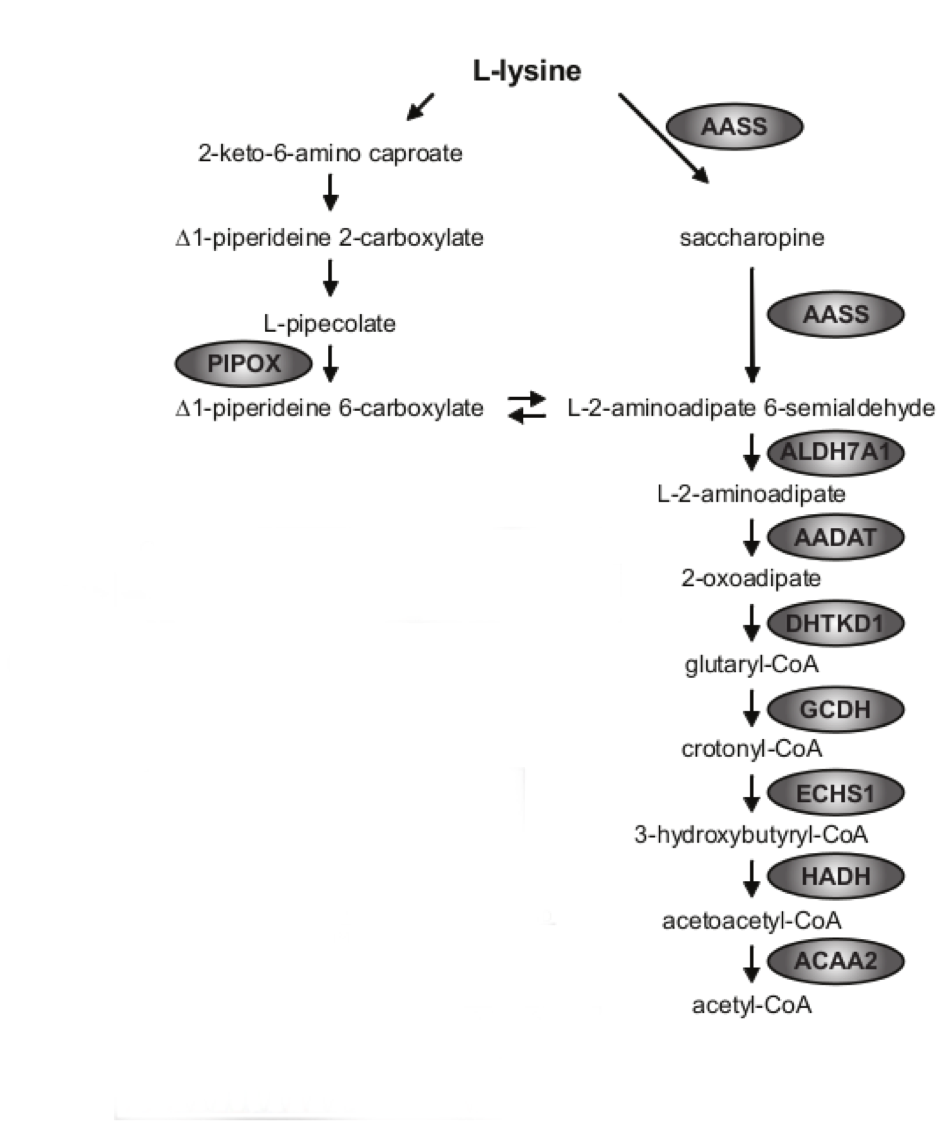
increased aminoadipic acid semialdehyde (AASA), increased pipecolic acid
Pyridoxine Dependent Epilepsy (aminoadipic acid semialdehyde dehydrogenase)
ALDH7A1 (antiquitin)
- High P6C complexes with pyridoxal 5 phosphate (B6)
Tx: B6, folinic acid, lysine restriction, arginine supplement
DDx; PNPO deficiency
Neuroimaging for GA1
Subarachnoid collections, Sylvian fissure enlargement, BG strokes
GA1 treatment
Limit substrate: Lysine free, reduced tryptophan; carnitine (glutaryl carnitine can be excreted), ?add arginine (compete with transporter)
ACP: C5DC
Low carnitine
UOA: 3-OH glutaric acid, glutaric acid
GA1
Clinical: Macrocephaly, encephalopathy, strokes (up to age 6, natural flux of transporter decreased as brain matures)
Gene: Glutaryl-CoA dehydrogenase (GCDH)
UOA: glutaric acid, 3-OH Glutaric acid (co-elutes with 2OH-GA) Decreased carnitine
ACP: glutarylcarnitine = C5DC LOW EXCRETERS EXIST
DDx for elevated glutaric acid in urine
gut bacteria overgrowth
ketosis
SCHAD (breaks down 3OH-glutaryl-CoA),
mitochondrial,
2OH-GA in GA2,
benign GA3 (NO C5DC),
renal disease,
maternal GA1
DDx for high glycine
NKH
VPA treatment
Ketotic (PA, MMA, IVA, B-ketothiolase deficiency)
PNPO deficiency
HIE (BBB breakdown)
prolonged fasting
Lipoic acid dependent pathways
BCKDH, PDH, 2-KGDH, 2-OADH, GCS (glycine cleavage system)
AA (CSF and Plasma) : Glycine elevated, Glycine CSF/Plasma ratio > .08
NKH
Genes: glycine decarboxylase/dehydrogenase (GLDC), aminomethyltransferase (AMT), Modified lipoic acid/dihydrolipoyl dehydrogenase (GCSH)
Elevated lactate in variant forms (lipoic acid/iron-sulfur cluster disorders)
NKH treatment
Na-Benzoate - conjugate with glycine to form hippurate which can be excreted;
dextromethrophan/ketamine for NMDA antagonism; folinic acid
Cherry Red Spots
NP-A (HSM)
GM1 gangliosidossis (HSM)
GM2 gangliosidosis (no HSM)
Sialidosis
Krabbe
Farber
Metachromatic leukodystrophy
Gaucher therapies
ERT: Imiglucerase, velaglucerase, taliglucerase
SRT: miglustat, eliglustat
Fabry therapies
ERT: agalsidase-beta, agalsidase-alpha
Chaperone: migalastat
Angiokeratomas
Fabry (a-Gal) - + renal/cardiac/stroke
Fucosidosis (a-fucosidase) - FUCA - + MPS features
B-Manosidosis (ID) - MANBA - + ID/neuropathy
Schilder (NAGA - aka a-GAL-B) - +neurodegeneration
Extensive mongolian spots
MPSI, MPSII, GM1
Heparan sulfate
Heparan = Head
MPS I, II, III, VII (Brain involvement)
Dermatan sulfate
Dermatan = bone + systemic
MPS I, II, VI, VII (Bone)
Keratan sulfate
MPS VI (Bone)
A-L-Iduronidase deficiency Treatment
MPS1 Hurler
HSCT before age 2
ERT: Iaronidase
Distinguishing features of MPSII vs MPSI
X linked
No corneal clouding
Dermal pebbling
Iduronidase 2- sulfatase
Iduronidase Sulfatase def. treatment
MPS II - Hunter
ERT: Idursulfase
MPSIII diagnosis
Urine: Heparan sulfate
Gene/enzyme:
Heparan N-Sulfatase (SGSH)
Alpha-N-acetyl-glucosaminidase (NAGLU)
aceyl-coa-glucosaminide acetyltransferase (HGSNAT)
N-acetylglucosamine-6-sulfatase (GNS)
MPS IV diagnosis
Morquio
Urine: Keratan sulfate
Normal intellect
Enzyme:
N-acetylgalactosamine-6-sulfatase (GALNS)
Beta-galactosidase (GLB1, Same enzyme as GM1 gangliosidosis)
Morquio treatment
ERT: Elosulfase Alpha for type IV A