Homocysteinuria
Clinical features
Ix
Tx
Most common inborn error of methionine metabolism
Presentation:
Infancy – non-specific features
i. Failure to thrive
ii. Developmental delay
c. Diagnosis usually made >3 years – when ectopia lentis occurs
Eye
- Downward lens dislocation (ectopia lentis)
- Glaucoma, astigmatism, cataracts, retinal detachment, optic atrophy
Skeletal (resembles marfans)
- Tall stature
- Long limbs
- pacts excavatum
- Osteoporosis (early onset) and flattened vertebrae
- genu valgum
- crowded teeth
Neuro
- Dev delay
- Intellectual disability
- Psych/behavioural difficulties
- Seizures
- Extrapyramidal signs /dystonia
Fair features: hair, skin, blue eyes, malar flush
Risk of vaso-occlusive disease (thromboembolism) - main cause of death
Ix
- Incr total plasma homocysteine and methionine
- incr urine homocysteine
- low or absent cystine in plasma
- Genetics: mutation in CBS
Tx
- High dose vitamin B6 (pyridoxine) - dramatic improvement in those who are responsive
- Folate supplementation
- Dietary methionine restriction
- Cystine supplementation

Spot diagnosis:
Infant with seizures, ataxia, developmental delay, alopecia and brittle hair, angular stomatitis, eczema-like rash, hearing loss
Biotinidase deficiency
- an inherited disorder in which the body is unable to recycle the vitamin biotin
- diagnosed by measuring serum biotinidase activity (will be reduced)

Spot diagnosis:
tall stature, downward lens dislocation, ID vintage
Homocystinuria
Spot diagnosis:
Macula - cherry red spots on fundoscopy
Developmental regression
Lysosomal storage disorder
- Diagnosis with serum white cell (leucocyte) enzymes
Ddx
- Neiman Pick disease - infantile HSMegaly, FTT/poor weight gain, developmental regression after the 1st birthday +/- interstitial lung disease
- Tay Sachs (GM2 gangliosidosis type 1) - Ashkenazi Jews, infant onset with early neurological regression, macrocephaly, hypotonia, seizures
Spot diagnosis:
kinky hair, hypotonia, laxity of joints/ skin, dev delay
What causes this condition?
What other manifestations can it have?
Ix
Tx
Menke disease
- mutation in ATP7A gene which is involved in Cu transport around body
- Also can cause prolonged jaundice and osteoporosis and subdural haematomas
Ix: low levels of serum copper and ceruloplasmin; light microscopy of hair follice can be diagnostic; genetic testing for ATP7a mutation
Tx: injections of Cu
What is the major cause of death and morbidity in homocystinuria?
Thrombo-embolism
What is homocystinuria caused by?
Mutation in CBS – gene encoding cystathione beta-synthase
Dietiary amino acid intake contains methionine (essential aa) -> converted to homocysteine -> requires CBS to convert it to cystine as metabolite
- > accumulation of homocysteine and methionine
- > deficiency in cysteine
Cystinuria
Cause
Presentation
Diagnosis
Tx
• Aminoaciduria; Rare AR disease 1/7000
• Defective aa transportation in prox renal tubules -> reduced reabsorption of cysteine ->
RECURRENT RENAL STONES due to cysteine crystallization
• Excessive cystine in urine
Diagnosis: urine cyanide-nitroprusside (urine turns magenta colour)
Treatment
• Regular fluids (dissolves stones)
• Alkalinisation of urine with potassium citrate (melts stones)
• Chelation (D- Penicillamine or tiopronin bind with cysteine to increase reabsorption)
Sepsis presentation (feeding difficulties, vomiting, dehydration, hypoglycaemia) in neonatal period
With metabolic acidosis (widened anion gap)
And high serum ammonia
Organic acidemia - ddx:
a. Methylmalonic acidaemia (MMA)
b. Propionic acidaemia (PA)
c. Isovaleric acidaemia (IVA)
d. 3-methylcrotonylglycinuria (3-MCG)
e. GA-1
NOT urea cycle defect (also get high ammonia w encephalopathy but respiratory alkalosis and NORMAL glucose)
Ix and Treatment of organic acidaemias
Ix
- blood gas: metabolic acidosis w high anion gap (>20)
- high ketones
- high serum ammonia
- low BSL
- often pancytopenia from bone marrow suppression
- Elevated organic acids in urine
Tx
- Low protein diet (decr aa substrate)
- Enhance enzyme activity (biotin, b12)
- Carnitine supplementation (binds to organic acids to enhance urinary excretion)
Which organic acidaemia RARELY presents in neonatal period and features
microencephalopathic macrocephaly, dystonia,
subdural haemmhorages
Glutaric academia (GA1)
Children typically present with a similar clinical picture to sepsis and may have associated infection and fever. Investigations will reveal metabolic decompensation (in response to infection/surgery/trauma etc) with ketoacidosis, hyperammonaemia, hypoglycaemia, and encephalopathy
What is the role of the urea cycle and what enzymes are involved?
Pathway by which nitrogen (produced from amino acid catabolism) is converted to urea for excretion
Protein -> ammonia (CNS toxic) -> urea cycle -> urea (non-toxic, excreted)
Enzymes
- OTC deficiency (most common) -> decr plasma citrulline, incr urine orotic acid
- Carbamylphosphate synthetase (most severe, presents early in neonatal period and quick progression to seizures, coma, death) -> reduced plasma citrulline and normal urine orotic acid
- Arginosuccinate synthetase deficiency -> incr arginase
- Arginosuccinate lyase deficiency -> incr citrullin
- Arginase deficiency -> incr arginine
Causes of lactate elevation
- Organic acidaemia (metabolic acidosis with high ammonia)
- Glycogen storage disorder (t1, Van Gierke’s)
- Mitochondrial disorders (also have elevated lactate in CSF and elevater serum CK)
What is the most common urea cycle defect?
And what is this, how does it present?
OTC (ornithine transcarbamyase deficiency)
- > x linked deficiency of OTC enzyme which is a mitochondrial enzyme located in liver and intestine
- > causes ammonium to build up, binds with glutamate to form glutamine
Presentation
After onset of BM feeds encepaloapthy with feeding difficulty, vomiting leathargy and progression to seizures and coma.
Glutamine has osmotic effect -> cerebral oedema
Milder form will present older during episode of illness/catabolism with encephalopathy and elevated ammonia
Presentation of urea cycle defect
Key features on ix
Presentation
- presents in neonatal period (males) after starting milk feeds (protein intolerance)
- recurrent vomiting
- decr GCS
- lethargy
- coma
- acute/chronic encephalopathy
Examples
OTC
Classic citrullinaemia (also known as argininosuccinate synthetase deficiency)
Arginase deficiency
Argininosuccinate lyase (ASL) deficiency (also known as argininosuccinic aciduria)
N-acetyl glutamate synthetase (NAGS) deficiency.
Key features:
Often self-limitation of protein intake as a learned behaviour in these patients
a. High ammonia
b. Respiratory alkalosis (elevated ammonia causes respiratory depression)
C. Liver dysfunction
D. Normal BSL! Normal ketones
Zellweger syndrome
Genetics/cause
Presentation
MRI
Bloods
Prognosis
‘cerebro-hepato-renal syndrome’
- AR
- Mutations in multiple PEX genes associated with peroxisome biogenesis
- Unable to import proteins into peroxisomes efficiently
Heterogeneous presentation within first few days of life, essentially progressive deterioration of liver, kidney, brain with death ~6mo after onset
- Dysmorphic
- FTT, feeding difficulties
- neurological (seizures, severe mental retardation, hypotonic, seizures, brain malformations)
- eye abnormalities (corneal opacification, retinal dystrophy)
- Chondrodysplasia punctata: stippled appearance of epiphyses on Xray
MRI - unmyelinated white matter
Bloods: incr VLCFA
Prognosis
- death in infancy
Hallmark feature of peroxisomal conditions (in terms of ix for diagnosis)
Presentation is abnormal FROM BIRTH
Incr VLCFA
What conditions cause high ammonia
Urea cycle disorder (VERY high ammonia; normal ketones/glucose/lactate)
Organic acid disorder (mild-mod lactate elevation; low glucose, high ketones and lactate)
Fabry disease
What is it
What is it caused by?
Features
Treatment
Most prevalent lysosomal storage disorder
Caused by mutation in GLA gene - defective alpha galactosidase A enzyme -> defective metabolism of glycosphingolipid (lipid) causing build up within lysosomes
pneumonic - FABRY C
F-foamy urine
Angiokeratoma around lower abdomen and upper thighs (red spots under skin), anhydrosis, alpha-galactosidase A deficiency
Burning pain in hands and feet with exercise, stress, illness (peripheral neuropathy)
Renal failure
Y chromosome - males (x linked), youth death
Cardiac disease, corneal clouding
If untreated -> cardiac and renal disease/failure
Tx: alpha galactosidase A enzyme replacement
Function of lysosome
- Digestive system of cell - degrades material from outside cell and digests obsolete cellular components
- Contains hydrolytic enzymes
Pancytopaenia and Hepatospenomegaly and recurrent fractures, most recently AVN of femoral head.
Blood Film shown below
What is this condition?

Gaucher disease
2nd Most common lysosomal storage disease (after Fabrys) caused by deficiency in enzyme glucocerebrosidase -> lipid accumulation within lysosomes in macrophages
Pathology - Gaucher cells look like crumbled tissue paper = lipid accumulation in macrophages
Hepatosplenomegaly (lipid fills liver and spleen)
Pancytopaenia (lipid fills BM)
B/L AVN femoral heads (reduced blood supply to bones, osteoporosis, fractures and pain crises)
+/- resp involvement (ILD, pulmonary vasc disease/pulm HTN)
+/- neurological involvement
—> type 1 no CNS involvement adolescent onset Ashkenazi jews
—> type 2 early CNS onset in neonatal period and death
—> type 3 chronic/later insidious onset)
Niemann Pick
What is it?
What is it caused by?
Sx
Lysosomal storage disease resulting in accumulation of cholesterol deposits inside lysosomes
NPC1 or 2 mutation -> impaired intracellular cholesterol transport -> brain, BM, liver, spleen, lung damage
Sx
- A characteristic early finding in children with NPC is impairment of the ability to look upward and downward (vertical supranuclear gaze palsy or VSGP
- hepatosplenomegaly
- prolongued cholestatic neonatal jaundice
- Thrombocytopenia secondary to splenic sequestration -> easy bleeding and bruising
Progressive neurological dysfunction
- Early infantile onset: delayed motor milestones, hypotonia, developmental regression
- Infantile and childhood onset: learning difficulties/ID, progressive cerebellar ataxia, dysarthria, dysphagia, seizures, cataplexia
- Teenage/adult onset: can be psychiatric sx, often misdiagnosed as early onset dementia
- Resp failure common reason for death
No cure
Tx is supportive
What are Mucopolysaccharidoses caused by?
What is the inheritance pattern?
Lysosomal storage disorders caused by a deficiency in enzymes required for breakdown of glycosaminoglycans (GAGs)
Fragments of partially degraded GAGs accumulate in lysosomes resulting in cellular dysfunction and clinical abnormalities
MPS 1-7
Range of clinical severity - Severe = Hurler syndrome, Mild = Scheie disease
All autosomal recessive EXCEPT MPS II which is X linked
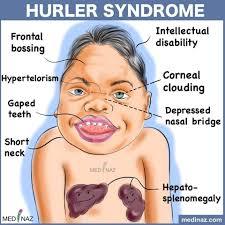
Clinical manifestations of MPS 1 (Hurler- Sheie syndrome)
What is it caused by?
Features?
Treatment?
Alpha L iduronidase deficiency (test for this enzyme in leukocytes and test urine for mucopolysaccharides)
- Dysmorphic features: Coarse features – wide nasal bridge, flattened midface, prominent forehead, coarse, thick hair, ‘puffy face, large tongue, prominent gums, large head
- Skeletal -short stature, kyphosis, stiff joints and contractures, Dysostosis multiplex (generalized thickening of most long bones, particularly the ribs)
- Eyes - corneal clouding, can being in 1st year of life and lead to blindness
Other:
Hepatosplenomegaly
Sinus Disease
Cardiovascular - valvular disease
Neuro- developmental delay, seizures, hydrocephalus
Soft tissue storage and skeletal disease with or without brain disease (MPS I, II (later onset), VII)
Soft tissue and skeletal disease (MPS VI)
Primarily skeletal disorders (MPS IV A and B)
Primarily CNS disorders (MPS III A to D)
Treatment
Enzyme replacement
BMT









-
Gastro242
-
Resp/ENT/Sleep240
-
ID287
-
Haematology185
-
Oncology163
-
Immunology194
-
Renal131
-
Endocrinology262
-
Cardiology294
-
Neurology168
-
Metabolics72
-
ECG23
-
Neonates114
-
Dermatoloy19
-
Rheumatology54
-
Genetics150
-
Emergency/surgical conditions141
-
Gen med/Dev med155
-
Psych/adolescents55
-
Pharmacology116
-
Epidemiology38
