2008 A Q1
Cerebral palsy is defined as a disorder of posture and movement due to an insult to the developing brain. What is the most common period for the responsible brain insult to occur?
A. Antenatal.
B. Intrapartum.
C. Neonatal.
D. 1 to 12 months.
E. After 12 months.
A. Antenatal.
What are the classic clinical features of infant botulism?
- Constipation
- cranial nerve abnormalities
- hypotonia
- hyporeflexia
- respiratory difficulties
other signs and symptoms
- poor feeding
- weak cry
- irritability
- lethary/decreased activity
What direction is the progressive weakness in infant botulism?
Descending.
Often starts with constipation several weeks prior to further symptom onset. Weakness then progresses over days.
Symptoms begin with cranial nerve involvement, loss of head control, weak cry, poor suck, impaired gag reflex, pooling of secretions and decreased oral intake.
What is the treatment of infant botulism and what should be avoided?
Treatment is supportive and should be commenced prior to confirmation of diagnosis. Diagnosis can take several days and the disease can progress within hours.
Respiratory and nutritional care should be commenced.
Avoid neuromuscular blocking drugs and aminoglycosides (gentamicin, tobramicin). Aminoglycosides kill the botulism and release further toxin.
What is the pathophysiology of infant botulism?
The anaerobic bacillus Clostrium botulin is ingested from either soil or honey. It colonises in the GI tract and produces a toxin absorbed by the terminal ileum.
Toxin binds to the presynaptic sides of the peripheral cholinergic synapses at ganglia and NMJs. Binding is irreversible and resolution of symptoms occurs when a new presynaptic terminal and synapse are sprouted. This takes approximately 6 months.
This patient also has recurrent sprained ankles and cramping in her hands.
What is the diagnosis?
What are other key clinical features of this disease?
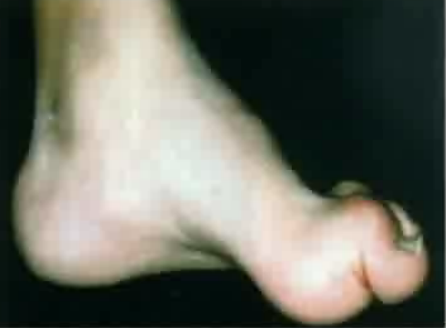
Charcot-Marie-Tooth disease is a demyelinating disorder of peripheral nerves. CMT1 is most common (40%) caused by duplication of PMP22 gene.
- Frequent sprained ankles caused by distal muscle weakness
- Loss of reflexes
- Pes cavus foot
- Hammer toes
- Distal calf muscle atrophy causing “stork leg” deformity.
- Clumsy walking due to motor and sensory loss
- Sensory loss gradual and mainly proprioception and vibration
Specific therapy not yet available, management is supportive.
Describe Guillain-Barré syndrome.
Guillan-Barr
- Most common cause of acute flaccid paralysis in infants and children.
- Group of disorders
- 2/3 have antecedant respiratory of GI infection.
- Campylobacter is most common pathogen.
- Neurologic symptoms occur 2-4 post an what appears to be benign infection.
Predominant symptoms = pain and gait difficulty.
Lower extremity symmetric weakness may ascend over hours to day to involve arms and muscles of respiration.
If cranial nerve involvement, usually CNVII.
Physical examination shows symmetric weakness with diminished or absent reflexes.
Nadir or function at 2-4 weeks, with return of function occuring slowly over weaks to months.
Diagnosis
- LP after 1 week of symptoms - Normal pressure, <10 cells with raised protein (>45gm/dL)
- MRI with contrast - enhancement of spinal nerve roots and cauda equina.
- Nerve conduction studies
What is the cardinal feature of myasthenia gravis?
Fluctuating skeletal muscle weakness, often with true muscle fatigue.
Myasthenia Gravis clinical features
Autoimmune disorder of neuromuscular transmission. Autoantibodies attack acetylcholine receptor (AChR), fix complement and decrease number of AChRs over time.
Uncommon - 10-20 new cases per million.
Bimodal ages of onset, peaks at 2nd-3rd decade (female predominance) and 6-8th decade (male predom)
Clinical features
Cardinal - fluctuating skeletal muscle weakness, often with true muscle fatigue (fatigue is worsening contractile force, not sensation of tiredness)
Weakness fluctuates throughout the day, usually worse evening/night or after exercise.
Look for:
- ocular - ptosis, eyelid fatigue, oculomotor paresis. Often unilateral or asymmetric. Historical alternating from side to side nearly always ocular myasthenia gravis.
- No pupil abnormalities.
- Bulbar - Fatiguable chewing, dysarthria, dysphagia
- Facial - appears expressionless, lost smile/horizontal smile
- Weakened neck and proximal muscles
- Respiratory muscle weakness may lead to myasthenia crisis.
Myasthenia Gravis diagnosis
- Acetylcholine receptor antibodies
- MuSK antibodies
- Repetitive nerve stimulation
- Single fibre electromyography
Duchenne and Becker Muscular Dystophies
Pathogenesis and Clinical Features
X-linked recessive.
Caused by mutations of the dystrophin gene on X chromosome (usually deletions)
Principipal symptom is weakness due to muscle fibre degeneration.
Pathogenesis - Dystrophin provides mechanical reinforcement to the sarcolemma and stabilises the glycoprotein complex, shielding it from degradation. Without dystophin, the glycoprotein complex is digested by proteases. This initiates degeneration of muscle fibres resulting in weakness.
DMD
1.3-1.8 per 10,000
Weakness
- onset of weakness at 2-3 years, proximal before distal, lower before upper limbs.
- difficulty running, walking, steps
- Gower’s sign (using hand to get up from floor)
- Often wheelchair bound by age 12
Cardiac
- Primary dilated cardiomyopathy and conduction abnormalities
Orthopaedic
- Arm and leg #s frequent
- Scoliosis
Examination Findings
- Pseudohypertrophy of calf and quads
- lumbar lordosis
- waddling gait
- shortening of achilles
- hyporeflexia or areflexia
Becker Muscular Dystrophy
Cf DMD, age of onset of symptoms later and less severe.
Mental retardation and contractures less severe.
Often remain ambulatory until age 15 years and often into adult life.
Prognosis
Pts with DMD often confined to wheelchair by age 12 yrs and die in late teens or twenties from resp insufficiency or cardiomyopathy.
BMD usually survive beyond 30 years.
Lab and Path findings in Duchenne and Becker muscular dystrophies
- Elevated CK (10-20 x upper limit of normal)
- ECG abnormalities
- tall right precordial R waves with increased R/S ratio
- deep Q waves in leads I, aVL, and V5-6
- Supraventricular arrhthmias in DMD
- EMG - myopathic changes
- Muscle biopsy - degeneration, regeneration, hypertrophic fibres, significant replacement of muscle by fat and connective tissue
- Dystrophin analysis - almost complete absence
Spinal muscle atrophy
An autosomal recessive group of disorders (types 1-4) characterised by degeneration of anterior horn cells in the spinal cord and motor nuclei in the lower brainstem.
SMA type 1 (infantile) is most common and most severe.
Presents in neonatal period, progresses quickly and most die before age 12 months from resp failure.
SMA 2 present b/w 3 and 15 months
SMA 3 (least severe) presents after one year
SMA 4 is adult onset, similar to SMA3. Presents 2nd-3rd decade.
Manifestations
- Diffuse proximal muscle weakness worse in lower limbs than upper.
- Absent or markedly decreased deep tendon reflexes
- SMA1 - severe symmetric flaccid paralysis, unable to sit unsupported
- Arthrogryposis (multiple joint contractures) may be present if prenatal onset
- Typically alert expression, normal eye movements
- Weakness of bulbar muscles = pooling of secretions, weak cry, poor suck and swallow, aspiration, tongue fasciculations
- Restrictive, progressive respiratory insufficiency
Diagnosis by muscle biopsy
What is Rett syndrome?
A neurodevelopmental disorder that occurs almost exclusively in females.
- epilepsy
- regression of development
- hand wringing
- ataxic, broad based gait
- sudden cardiac death
Development is normally initially.
At 12-18 months there is lost of acquired fine motor, intellectual and communicative skills.
Regression can be rapid, or slowly progressing.
There are stereotypical hand movements which are incessant when awake, but cease during sleep (rubbing, clapping, kneading, pill rolling, hand opposition etc)
Broad based, ataxic, clumsy gait. Sometimes difficulty crossing from one type/colour floor surface to another.
Scoliosis in 50-85%
Which anticonvulsant requires close serial measurements of serum levels due to narrow therapeautic index?
phenytoin
In the hand, what do the ulnar, median and radial nerves supply?
Ulnar N -
- motor - intrinsic muscles of hand
- sensation - lateral fingers to lateral half of ring finger
Median N -
- motor - thumb adductors
- sensation - pointer and middle fingers to half of ring finger
Radial
- nil motor
- sensation - posterior thumb web
Transverse myelitis
Transverse myelitis is characterised by rapid development of motor and sensory deficits.
Presents with discomfort/pain at the level of the lesion and progresses to numbness and weakness in the truncal and appendicular musculature.
Paralysis begins as flaccidity and over several weeks spasticity develops.
Urinary retention is an early finding with incontinence occurring later in the course.
Diagnosis made on the basis of clinical findings (bilateral sensorimotor and autonomic spinal cord dysfunction, clearly defined sensory level, progression to nadir of clinical deficits between 4h and 21 days after onset of symptoms), demonstration of spinal cord inflammation (CSF pleocytosis or MRI with gadolinium enhancing cord lesion) and exclusion of compressive, post-radiation, neoplastic and vascular causes.
Recovery is likely to be complete in older children but in younger children (<3 years), neurological recovery is often incomplete with only 40% likelihood of independent ambulation.

What is the cellular action of botulinum neurotoxin?
Binding – toxin attaches rapidly and avidly to the presynaptic nerve membrane
Internalisation – toxin crosses the presynaptic membrane without causing onset of paralysis
Inhibition – toxin inhibits the release of acetylcholine, diminishing the endplate potential and causing impaired neuromuscular and autonomic transmission
Recovery of impulse transmission occurs gradually as new nerve terminals sprout and contact is made with the post-synpatic motor endplate (takes 6- 8 weeks)
A 5 month old boy with congenital heart disease is evaluated because of motor delay.
Diagnosis?
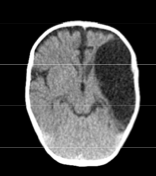
Arachnoid Cyst
CT Scan demonstrates a large fluid collection in the left parietal-temporal region. The fluid is the same density as CSF. This scan and history is most consistent with an arachnoid cyst.
The brain tissue looks relatively normal which would make cerebral malformation less likely.
Blood is of a higher density than CSF (acute blood is white, chronic looks slightly darker than brain tissue)
Anterior Horn cell disease is a form of SMA. How does it often present?
Because anterior horn cell disease affects the nerve cells responsible for body movements, precursors to the disease include severe reduced muscle tone, diminished limb movements, lack of muscle reflexes, body tremors, difficulty eating and drinking, breathing problems, or inability to stand.
Look for tongue fasciculations
What side-effects are common to most antiepileptic medications?
Drowsiness, ataxia, tremor, nystagmus, dysarthria, confusion, nusea, vomiting, sleepiness or insomnia, mood disturbance.
What are the idiosyncratic side effects of carbamazapine?
- rash
- leukopenia
- hyponatremia
- irritability
- weight gain
What are the idiosyncratic SEs of lamotragine?
- Rash
- severe hypersensitivity syndrome
What are the idiosyncratic SE’s of phenytoin?
- Rash
- Serum sickness type illness
- Extravasation









-
Genetics54
-
Haematology33
-
Oncology35
-
Neonates32
-
Nephrology36
-
Cardiology58
-
Respiratory43
-
Neurology51
-
Developmental65
-
Emergency42
-
Gastroenterology52
-
Endocrinology34
-
Pharmacology31
-
Microbiology16
-
Evidence Based Medicine6
-
Immunology15
-
Infectious Diseases30
-
General2
-
Dermatology37
-
Metabolic11
-
Mental Health2
-
Rheumatology2
-
Cardiology Exam Qs 2004-199972
-
Neurology Exam Questions83
-
Gastroenterology Exam Questions60
-
Cardiology Exam Q's 2008-200549
-
Statistics and Ethics Exam Questions20
-
2008 Paper A70
-
2008 Paper B100
-
2010 Remembered Paper8
-
2013 Remembered86
-
2012 A Remembered69
-
2012 B Remembered89
-
2007 Paper A70
-
2007 Paper B100
-
2011 A Remembered63
-
2009 Remembered4
-
2011 B Remembered89
-
2006 Paper A70
-
2006 Paper B100
-
2005 Paper A70
-
2005 Paper B35
