Insulinfrage, welche Applikationsarten gibt es, supramolekulare und molekulare Unterschiede
Insulinfrage auf supramolekularer, molekularer, galenischer und applikativer Ebene
Erklären Sie am Beispiel von Insulin die Möglichkeiten der verzögerten Liberation bei parenteral applizierten Formulierungen!
Was sind Basalinsuline, wie aufgebaut & worauf basiert die lange wirkdauer ?
Verschiedene Type bezogen auf die Liberation von Insulin aufzählen und beschreiben mit Beispiel
-Insulin kann s.c. (=Standard), i.m. (30-50% schnellere Wirkung) oder i.v. (nur unter äußerster Vorsicht–> hypoglykämischer schock) gespritzt werden
Schnell wirksames Insulin: Analoginsuline, Bolusinsuline
- Molekulare Ebene
- Wirkungseintritt nach 0-15min
- Wirkdauer 2-3h
- Z.B. Insulin Lispro (Prolin und Lysin gegeneinander ausgetauscht), hat gleiche Bindungsaffinität am Rezeptor wie Normalinsulin, –> Geringer selbstassoziationn (schnellere Zerfall von Hexameren & Dimeren)
Kurz wirksames Insulin: Normalinsulin, Bolusinsuline
- Molekulare Ebene
- Wirkungseintritt nach 15-30min
- Wirkdauer 4-6h
Intermediärinsuline: Semilente, Basalinsuline
- Supramolekulare Ebene
- Amorphe Form
- Wirkungseintritt nach 30-90min
- Wirkdauer 10-24h
Zink-verzögert
- amorphe Zinkinsuline supramolekular
- Zusatz von Zinkkonzentrationen
Protamin-Zink-Insulin
- enthält großen Protamin-Überschuss
- Insulin bildet mit Zink-Ionen einen Komplex –> bei pH-Wert des Gewebes schwer löslich –> verzögerte Resorption des Insulins = Supramolekular
Surfen-Insulin
- Komplexbildung mit Aminoquinurid –> supramolekular
- bei pH 3,5 gelöst, fällt noch Applikation im neutralen Gewebe amorph aus
Langzeitinsuline: Ultra-Lente, Basalinsuline
- Teilweise supramolekulare, teilweise molekulare Ebene
- kristalline Form
Zink-verzögert
- gezielte Kristallisation in Anwesenheit hoher Zn-Konz.
- Supramolekulare Ebene
Langwirksames Insulinanaloga
- Molekulare Ebene
- durch Modifikation der AS-Ketten verzögert abgebaut
- z.B. Insulin glargin (Lantus) & Detemir (Levemir)
- L_antus –> durch AS Tausch & verlängerung –> saure klare Lösung falleb bei s.c. appl. schlecht lösliche Mikropräzipate aus (Mikrokristalle)_
- Detemir zustäliche Fettsäure (Myristinsäure) angebaut, die an Albumin bindet –> Verzögerung
Einfluss Partikelgröße und Kristallinität auf Resorptionseigenschaften (supramolekulare Ebene)
- amorpher Bereich: Wirkdauer deutlich verkürzt, aber höhere Konzentration
- Kristalliner Bereich: Wirkdauer lang, geringere Konzentration
–> Insulinpumpen CSII (continuous subcutaneous Insulin Infusion), Pumpe tragbar am körper, Normalinsulin mit Stabilisatoren PEPG
–> Nicht invasive injektion Jet-Injector –> injektion von flüssigem lyophiliserten ASt, ohne Nadel mit Druck in die Haut (Pulverinjektion)
–> Inhalator –> pulmonal Applikation konnte sich nicht durchsetzen

Definition
a) delayed, prolonged
b) housekeeper Phase
c) Schmelztabletten
d) implantierbares Therapeutisches System
nüchtern Einnahme
(plus wollte er bei jeder Definition noch ein Beispiel dazu)
e) Peyer Plaques
Was bedeutet: Einnahme nach der Mahlzeit, während der Mahlzeit, zwischen den Mahlzeiten?
Delayed = verzögerte Freisetzung, Magensalresistente Arzneiformen, keine Retardarzneiform; der Wirkungseintrig erfolgt zeitlich verzögert • Nüchterne Einnahme empfohlen, da es hier ansonsten zu langen Verweilzeiten im magen kommt, weil sie erst bei der nächsten housekeeper Phase weitertransportiert werden
Prolonged = verlängerte/anhaltende Freisetzung 1.Ordung , Retardarzeiformen; die Wirkung ist verlängert im vergleich zu einer schnell freisetzenden AF
• Meist einnahme mit oder nach Essen gut, da sich so die Wirkung verlängert
Housekeeper Phase:
3te Phase im interdigestiven Zustand, trig nach ca. 1,5 h im nüchternen Zustand ein. In dieser Phase kommt es zu starken gleichmäßigen Kontraktionen, die Partikel über 2 mm aus dem Magen enxernen –> AF, die im Magen nicht zerfallen sind hier dabe
Schmelztablegen
= schnell freisetzende Tablegen, die zum auflösen im Mund behalten werden und dort sehr schnell zerfallen bevor sie geschluckt werden. Oft Lyophilisate Vorteile: hohe Lagerstabilität, schnelle Freisetzung Ideal für Patienten mit Schluckbeschwerden Z.B. Levetiracetam
• Einnahme nicht mit dem Essen, ob nüchtern oder nicht nüchtern egal, da es im Mund resorbiert wird bzw. schon gelöst im Magen ankommt und nicht über 2 mm groß ist
Implantierbares Therapeutisches System
- Implantierbare Pumpensysteme und therapeutische Systeme
- Implaniterbare Polymere
- Keimfreie Depotarzneiformen, die entweder durch chirurgischen Eingriff oder mit einem Injektor in das unterhautgewebe implan9ert werden
- Zylinderförmige, starrte Polymerstäbchen mit eingebegetem Wirkstoff
- Freisetzung aus Polymeren durch Diffusion oder Matrixerosion
- Z.B. Alzet
Peyer Plaques
- Spezielle Zellsysteme im GIT
- Lymphatisches Gewebe im GIT : Peyer´sche Plaques im Dünndarm (v.a. Krummdarm), Lymphfollikel im Dickdarm (Rektum)
- Rolle –>:
- Immunabwehr, Aufnahme von Antigen (Impfstoffe!)
- Resorption von Mikropartikeln (< 10 µm), Makromolekülen
- Neue Drug Delivery Ansätze

Therapeutische Systeme: Beschreibung von zwei und Funktion durch Skizze erklären
Was ist ein orales therapeutisches System, welcher Freisetzung folgt es, Kinetik?
OROS erklären
Definition orales osmotisches therapeutisches System?
Implantierbare
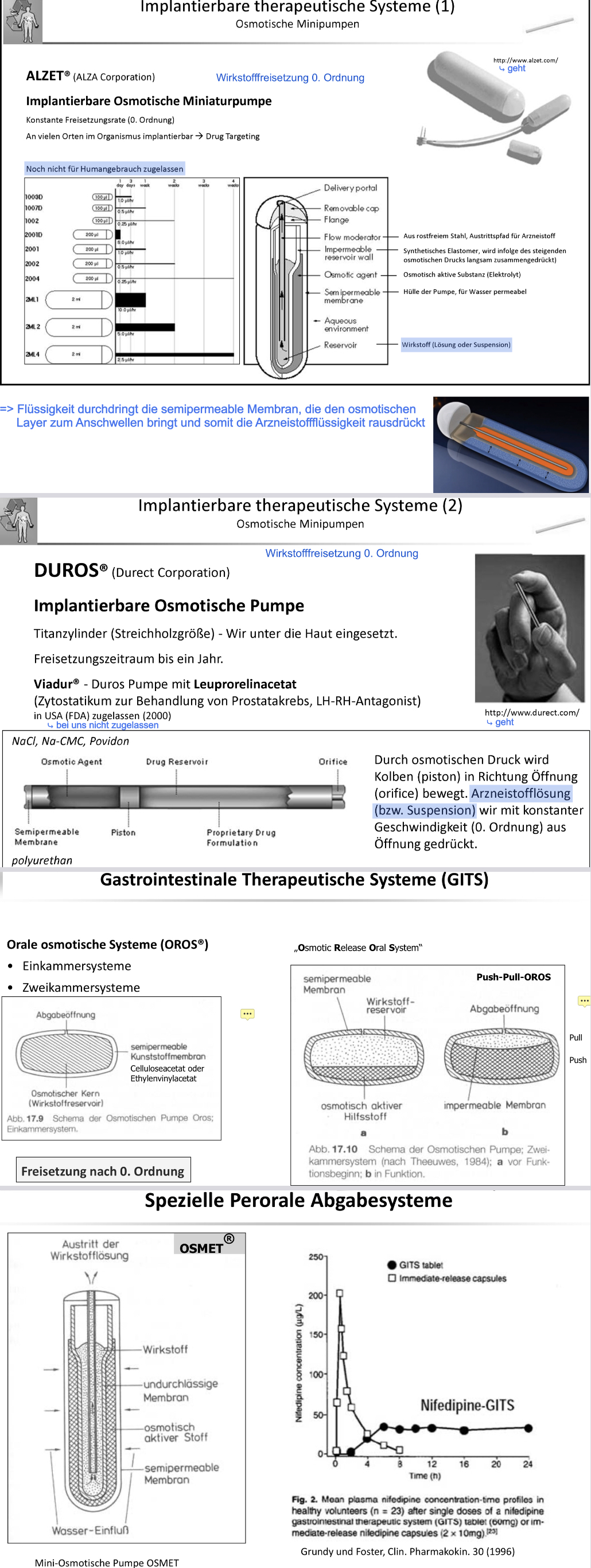
I. ALZET
Art Kapsel als Morphologie
Der Äußere Teil ist ein Polymer (Membran) was für Wasser durchlässig ist aber nicht für Elektrolyte (Elektrolyt Typisch wären Natriumchlorid, Kaliumchlorid, Treibmittel), die dann den entsprechenden osmotischen Druck aufbauen wenn sie mit Wasser in Berührung kommen. Durch diesen osmotischen Druck (der gesteuert wird wie langsam Wasser durch diese Membran eindringen kann, bildet sich in der Äußeren Schicht ein Überdruck und dieser Überdruck quetscht den inneren Teil (der mit einem Gel gefüllt ist) zusammen und drückt den Wirkstoff aus der Kanüle
–> Drug Targeting
II. DUROS
siehe bild
-ORALE THERAPEUTISCHE SYSTEME (OROS)
ADALat
• Spezielle Perorale Abgabesysteme
(–> Arzneiform zur langanhaltenden, gleichmäßigen Freisetzung eines WS. Die Freisetzung wird hierbei vom osmotischen Druck kontrolliert)
A) Einkammersysteme:
B) Zweikammersysteme:
Vorteil:
gelichmäßiger Plasmaspiegel
Resorption unbeeinflusst von Nahrungsaufnahme &pH
Nachteile
aufwendige Produktion.
die Tablette enthält genug Wirkstoff für 24 Stunden und ist deshalb ziemlich groß. …
bei Wirkstoffen mit einer hohen Halbwertszeit (>2 Stunden) ist eine Initialdosis notwendig.
da die Arzneiform nicht zerfällt, wird sie über den Stuhl ausgeschieden.
Vor- und Nachteile von Implantaten
Vorteile
- Konstante, verlässliche Blutspiegel über lange Zeit
- Keine Einnahme zu bestimmten Zeiten notwendig (gute Compliance)
- Gesteuerte Freisetzung nach zirkadianem Rhythmus
- Einstellbarkeit der Freisetzungsrate
- Depotsysteme für systemische Langzeittherapie, lokalisierte Depotsysteme (Infektions-, Krebstherapie)
Nachteile
- Chirurgischer Eingriff
- Nicht abbaubare müssen wieder entfernt werden
- Hohe lokale Wirkstoffkonzentration
- Hohe Entwicklungskosten

Was ist der scheinbare Permeationskoeffizient. Berechnung! Ab welchem Schwellenwert spricht man von guter Permeabilität?
Was ist der scheinbare Permeationskoeffizient, wie wird er eingeteilt in hoch mittel und niedrig, Formel?
Scheinbarer Permeationskoeffizient, schwellenwert für hohe permeation
Papp Formel & Bezeichnungen
=Maß für die Geschwindigkeit, mit der eine Substanz unter Steady-State Bedingungen permetiert
–> hoher Wert = höchst wahrscheinlich keine Probleme bei der Resorption

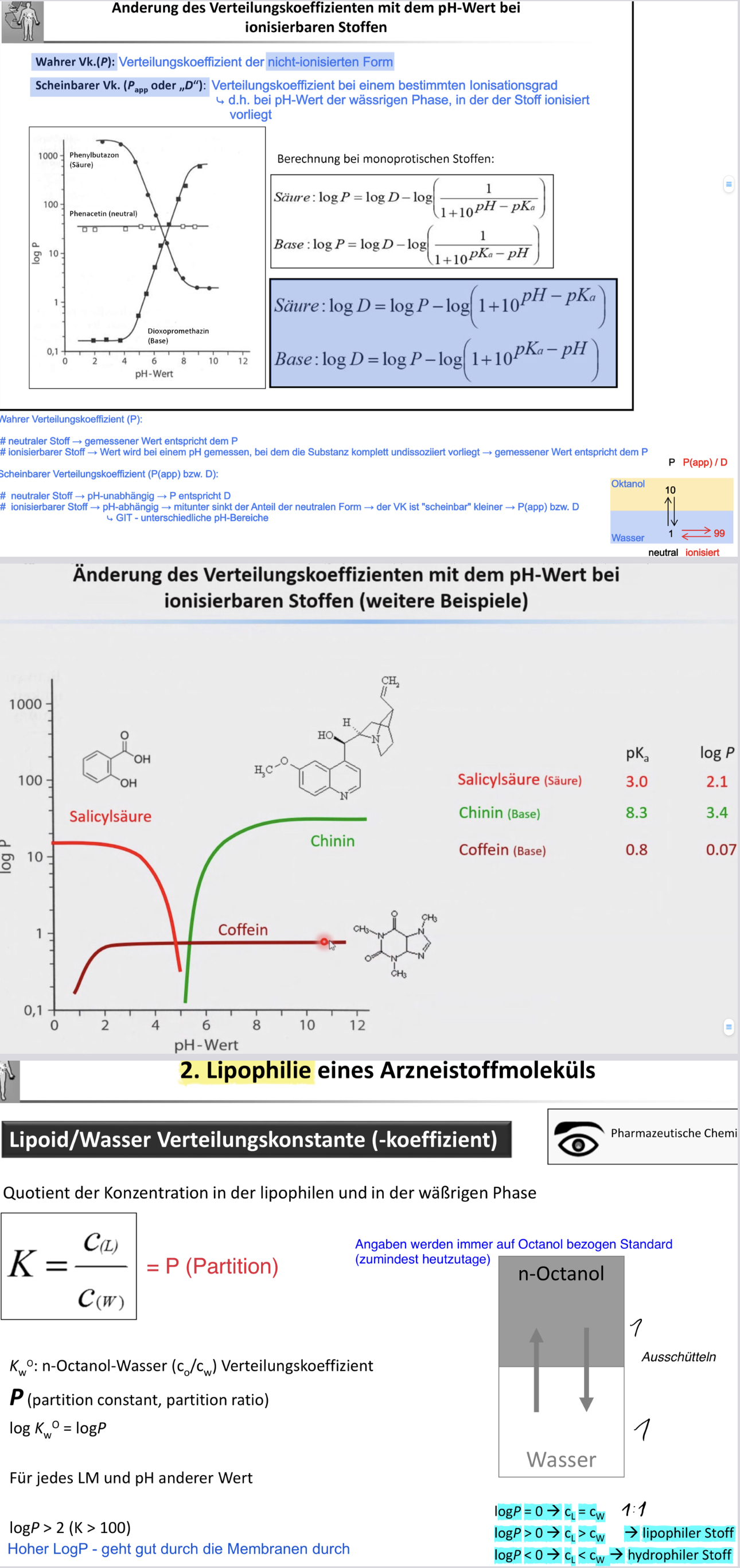
Wie hängt bei einem monoprotischen Stoff die Verteilung PH-Wert ab/Erklären Sie den Zusammenhang zwischen PH-Wert und Verteilungskoeffizient?
Berechnen Sie den log D eines basischen Arzneistoffes mit einem log P von 3.7; bei einem ph von 3,8 und einen pka von 6,2
Scheinbaren Verteilungskoeffizient-zusammenhang mit Säure/ Base angeben und Zusammenhang zwischen Octanol/Wasser und Sättigungslöslichkeit
Wahrer Vk(P) = Verteilungskoeffizient der nicht ionisierten Form
Scheinbarer Vk(Papp oder „D“) = Verteilungskoeffizient bei einem bestimmten Ionisationsgrad (d.h. pH-Wert der wässrigen Phase, in der der Stoff ionisiert vorliegt.)
Der Verteilungskoeffizient ist bei nicht ionisierten Stoffen am größten, weil nur diese in der Lage sind, biologische Membranen zu überwinden (diese verhalten sich dann lipophil). Je nachdem ob es sich bei dem Stoff um eine Säure oder Base handelt, liegt dieser bei einem bestimmten pH- Wert ionisiert oder nicht ionisiert vor. Zum Beispiel liegt Salicylsäure bei geringem pH von ca. 0-3 protoniert und damit nicht ionisiert vor, kann somit Biomembranen (Basallöslichkeit) leicht überwinden und besitzt daher einen höheren Verteilungskoeffizienten. Steigt der pH- Wert jedoch an (ab ca. 4-5), so wird die Salicylsäure deprotoniert, liegt ionisiert vor (ist also hydrophil und leichter löslich) und kann daher Biomembranen nicht mehr überwinden, wodurch der Verteilungskoeffizient sinkt.
Allgemein:
Ph-Einfluss auf die Löslichkeit von monoprotischen Stoffen
Ein Stoff der protoniert/deprotoniert werden kann, kann ionisiert oder nicht ionisiert vorliegen. Besser löslich ist immer die ionisierte Form. Die nicht Ionisierte Form wäre lipophiler und könnte besser resorbiert werden.
pH hat einen sehr großen Einfluss, da er bestimmt, ob die Säure deprotoniert vorliegt oder die Base protoniert ist.
Basen sind im Sauren protoniert, Säuren im Basischen deprotoniert.
Wasser/Octanol Verteilungskoeffizient verwendetes als Standard Maß zur Beurteilung der Lipophilie und Membrangängigkeit von Arzneistoffen
hixson crowell, wieso Korrektur notwendig? welches Problem ergibt sich?
F (Oberfläche des Feststoffen), ist bei Verwendung von z.B Partikeln, Granulaten or Pellets etc. nicht konstant –> Näherung wäre Falsch, daher korektur da die Fläche während dem auflösen kleiner wird.
Problem:
Fläche F wird mit zunehmder Auflsöung des Stoffes kleiner –> Fläche konstanthalten ( kleiner Fläche würde Verlangsamung bedeuten) –> Roating Disk Apparatus(rotierende Tablette)

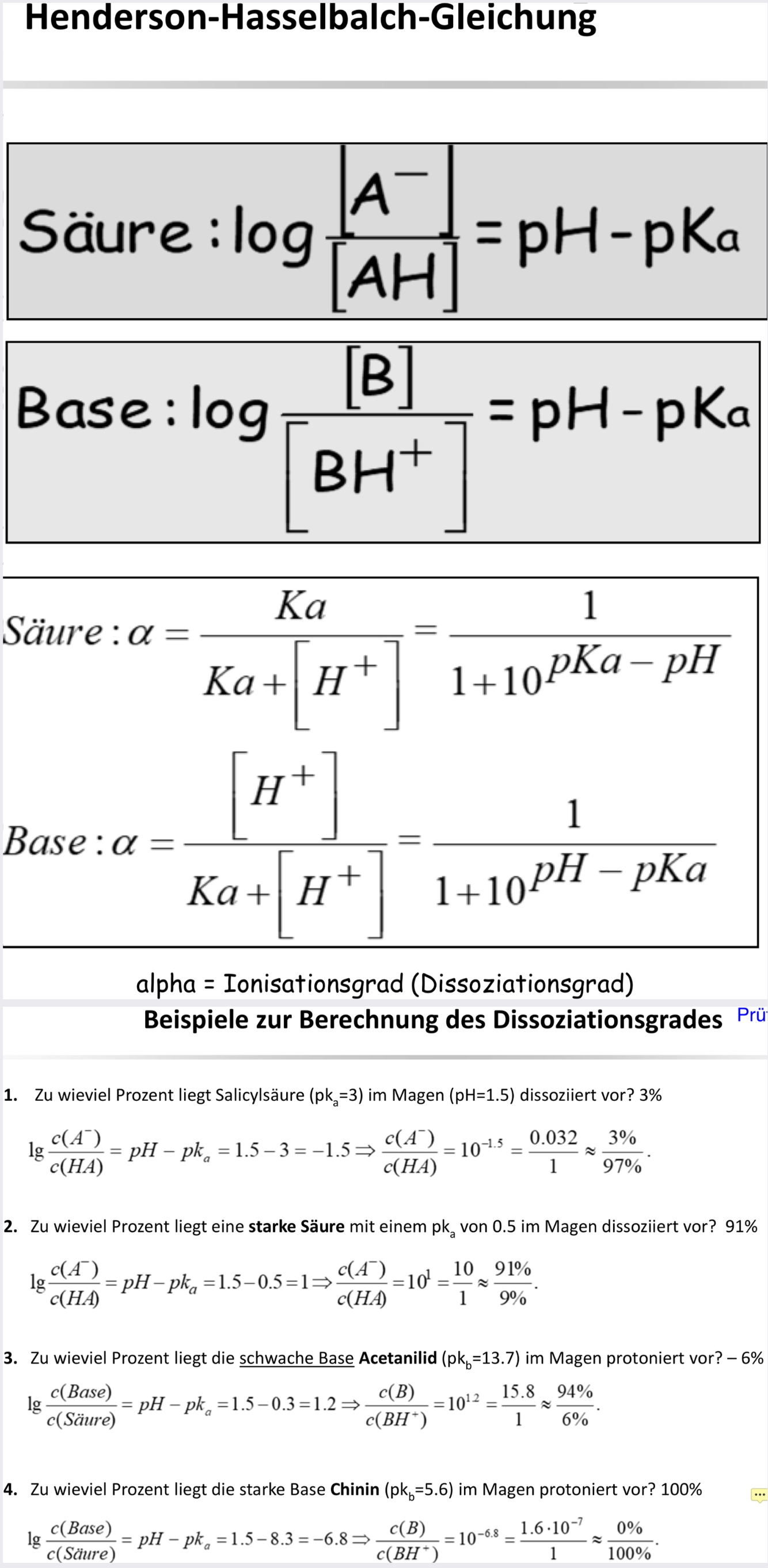
Rechnung mit pH und pkA, dissoziatsion % in Blut
.Dissoziationsgrad von Säuren und pKa im Blut
Berechnung wie viel Säure dissoziert
Berechne zu welchem Anteil dissoziiert Säure bei PH 7,7 und pka 6,8
Eine-Säure.mit-pKa-4,5-liegt-bei.pH.5,6 -zu:3%-dissozilert-vort
-Eine Säure hat einen pka wert von 4,5 bei einem ph wert von 3. Wieviel %
sin dissoziiert?
Dissoziierten Teil des ASt berechnen
- schwache Säuren sind 2 pH-Einheiten unterhalb ihres pKa praktisch vollkommen undissoziiert (pH=4, pka=6)
- 2 pH-Einheiten über pka völlig dissoziiert (pka=6, pH=8)
- pH=pka: 50% des Ast. liegt dissoziiert vor
MERKE:
- hoher pka Base = starke Base
- hoher pka Säure = schwache Säure
- schwache Basen = optimale AM
Arzneistoffe werden vorwiegend in nicht-ionisierter Form resorbiert; dabei gilt: Säuren sind im Sauren gut resorbierbar, Basen im Basischen.
Merke: bei starken Säuren und starken Basen liegt das Gleichgewicht immer auf der ionisierten Seite;
Berechne zu welchem Anteil dissoziiert Säure bei PH 7,7 und pka 6,8
- Hier säure deshalb log (A-(AH) = pH-pKa –>1/(1+107,7– 106,8)
Zu wie viel% ist Säure (pKa=5,4) im Blut (pH=7,4) dissoziiert?
- log (Salz/Säure) = pH-pKa
- 7,4-5,4 = 2 –> 102 = (100/1) = (99%/1%)
- –> Säure ist zu 99% dissoziiert
erkläre Verhältins Dosierungsintervall
Dosierungsintervall wann kumulation
t 1/2 >> τ
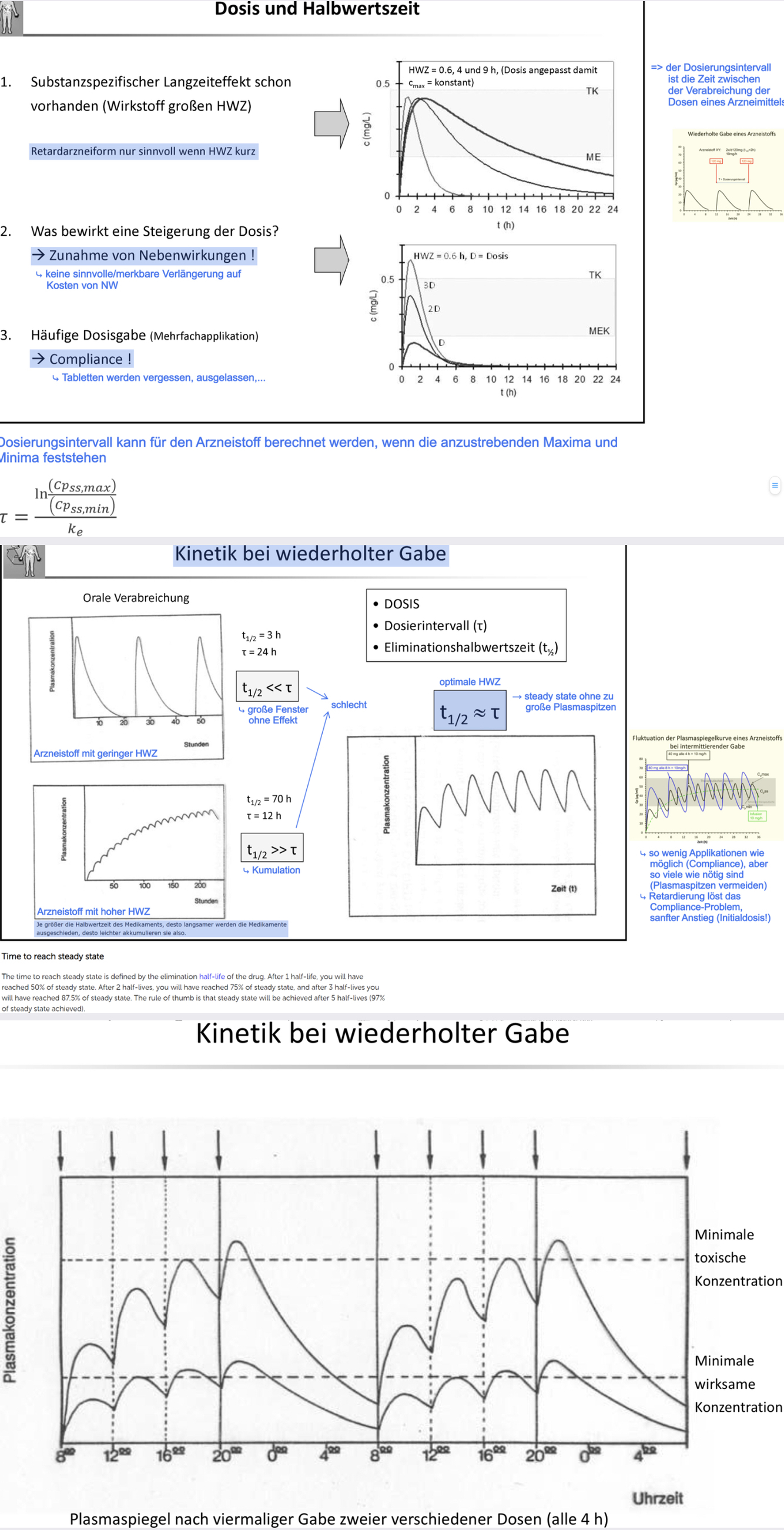
HWZ ist größer als die Einnahme –> Klassische Kumulation und wir landen im toxischen Bereich
Anhäufung von Arzneimitteln im Organismus -> wenn die Ausscheidung des Medikamentes geringer ausfällt als die Aufnahme t 1/2 >> τ
- Gefahr einer Überdosierung oder Intoxikation, vor allem bei Medikamenten, die über die Niere ausgeschieden und niereninsuffizienten Patienten verabreicht werden
- Zur Vermeidung einer Kumulation wird bei eingeschränkter Nierenfunktion die Dosis reduziert
- erfolgt immer dann, wenn die Halbwertszeit des Ast größer ist als das Dosierungsintervall
- Z.b. Gabe von Nitrazepam (HWZ 15-30 h) jeden Abend (d.h. Dosierungsintervall alle 24 h) als Schlafmittel würde zur Kumulation führen
- relatives Dosierintervall < 1
Erklären Sie
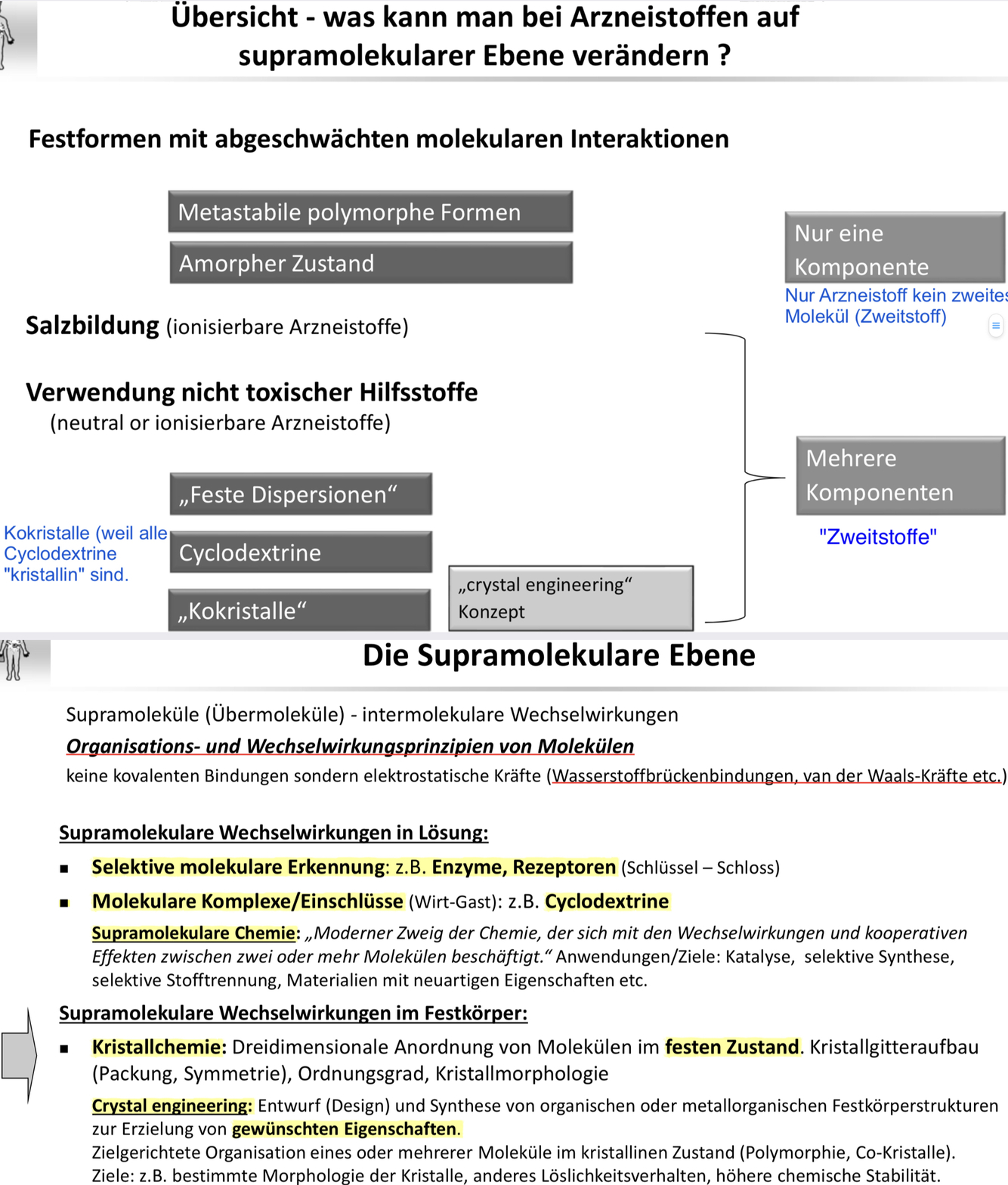
feste Dispersion,
Salz,
Kokristall,
Begründer der Biopharmazie, & deren aufgaben
Feste Dispersion
= Hochdisperse = Wirkstoff in (ultra-)feiner, hochdispersen Form im hydrophilen Hilfsstoffträger verteilt -> binäres System
➢ Matrix: entweder kristallin oder amorph
➢ AS: kann molekular dispers, in amorphen Partikeln oder in kristallinen Partikeln vorliegen -
Bsp.: Eutektika, Glassuspensionen, amorphe Präzipitat
Feste Lösung
molekulardisperse (feste Lösung) Verteilung
eines (schwer löslichen) Wirkstoffs in einem festen, hydrophilen (oft makromolekularen) Hilfsstoff (-träger).
= binäre Systeme (wen nur zwei Komponenten beteiligt sind)
Kristaline PEG
Amorphe PVP
Hilfsstoffe bzw. Trägerstoffe: Polymere (PVP, PEG), Zucker und Zuckeralkohole, Andere wasserlösliche Träger (Methylcellulose, HPMC, Zitronensäure, Harnstoff)
Kokristall
Mehrkomponenten-Kristalle - neutraler Wirkstoff + Zusatzstoff (Hilfsstoff) Kristallisation: Ko-Kristall
- durch Änderungen der intermolekularen Wechselwirkungen in der Kristallstruktur ändern sich auch die Eigenschaften des Feststoffes;
Polymorphie
Versch. Kristallstrukturen bei exakt gleicher chemischer Zusammensetzung der Kristallformen;
Salz
Verbindungen, die aus gleich vielen Kationen (positiv geladen) und Anionen (negativ geladen) bestehen haben daher keine Ladung. Die beiden Ionen ordnen sich in einer Gitterform, dem Ionengitter, an.

Problemstoffe bezüglich der Bioverfügbarkeit?
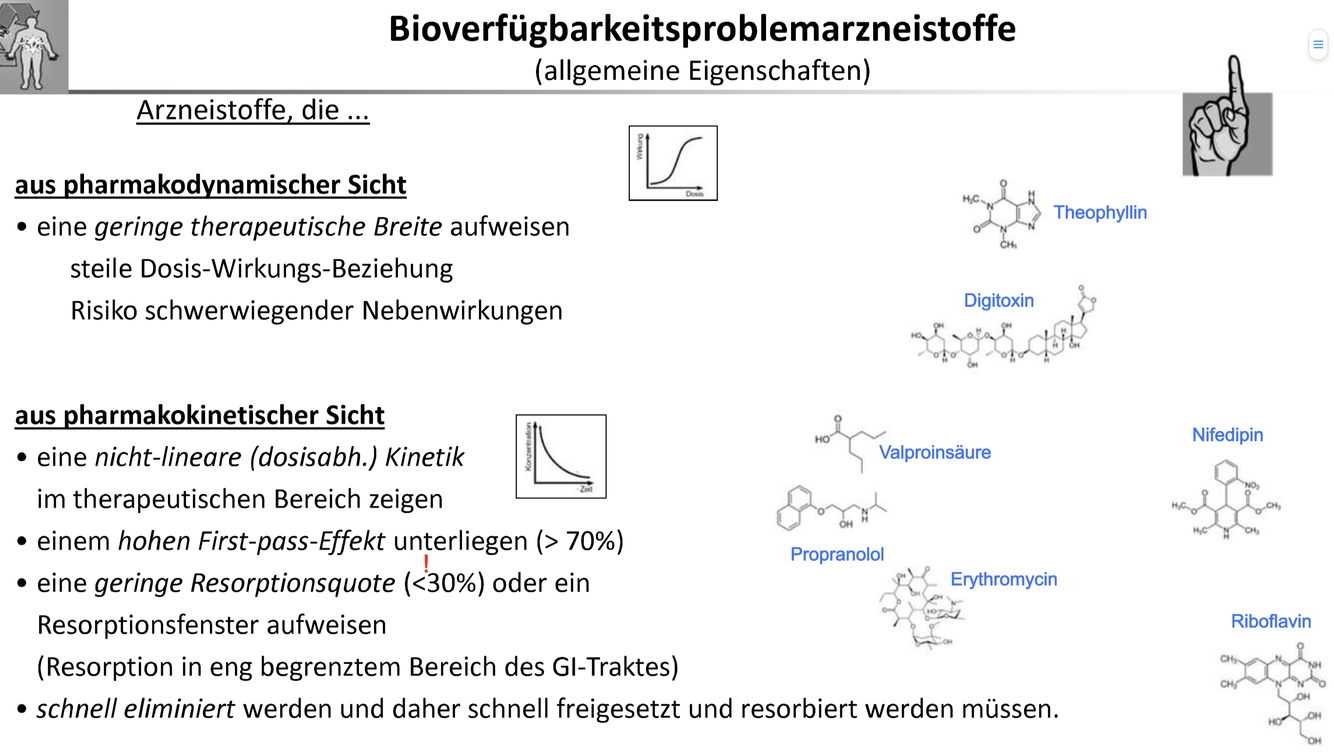
Bioverfügbarkeitsproblemarzneistoffe aus pharmakokinetischer Sicht
Folgende pharmakokinetischen Eigenschaften sind Bioverfügbarkeitsproblemarzneistoffe (4aufzählen) (??)
Welche eigenschaften sind kritisch hinsichtlich idealer Bioverfügbarkeit (Bioäquivalenz)
Aus physikalisch-chemischer Sicht: 4 stk
a) Aus pharmakodynamischer Sicht:
- Geringe therapeutische Breite, steile Dosis-Wirkungs-Beziehung
- Risiko schwerwiegender NW
b) Aus pharmakokinetischer Sicht:
- Zeigen eine nicht lineare (dosisabhängige) Kinetik im therapeutischen Bereich
- Einem hohen first-pass-Effekt (>70%) unterliegen (Propanolol)
- Eine geringe Resorptionsquote (<30%) aufweisen
- ein Resorptionsfenster aufweisen (Riboflavin)
- Schnell eliminiert werden und daher schnell freigesetzt und resorbiert werden (Erythromycin)
c) Aus physikalisch-chemischer Sicht:
- Eine schlechte Löslichkeit aufweisen(< 0.1% in Puffer pH 7 oder in 0.1 N HCl)
- Eine geringe Auflösungsgeschwindigkeit besitzen (< 50% in 30 min)
- Schlecht benetzbar sind oder die Partikelgröße kritisch ist
- Am Resorptionsort, bei physiologischen Bedingungen chemisch nicht stabil sind (saurer Mageninhalt)
- In verschiedenen Kristallformen vorliegen können
- Molekülgröße zu groß
- Sehr hohe Lipophilie/Hydrophilie
- Sehr stark sauer oder basisch
- Mehr als 5H-Donoren
- Mehr als 10H-Akzeptoren
- Sehr kurze HWZ
Beispiele Theophyllin, Erythromycin, Digitoxin, Valproinsäure, Propanolol….
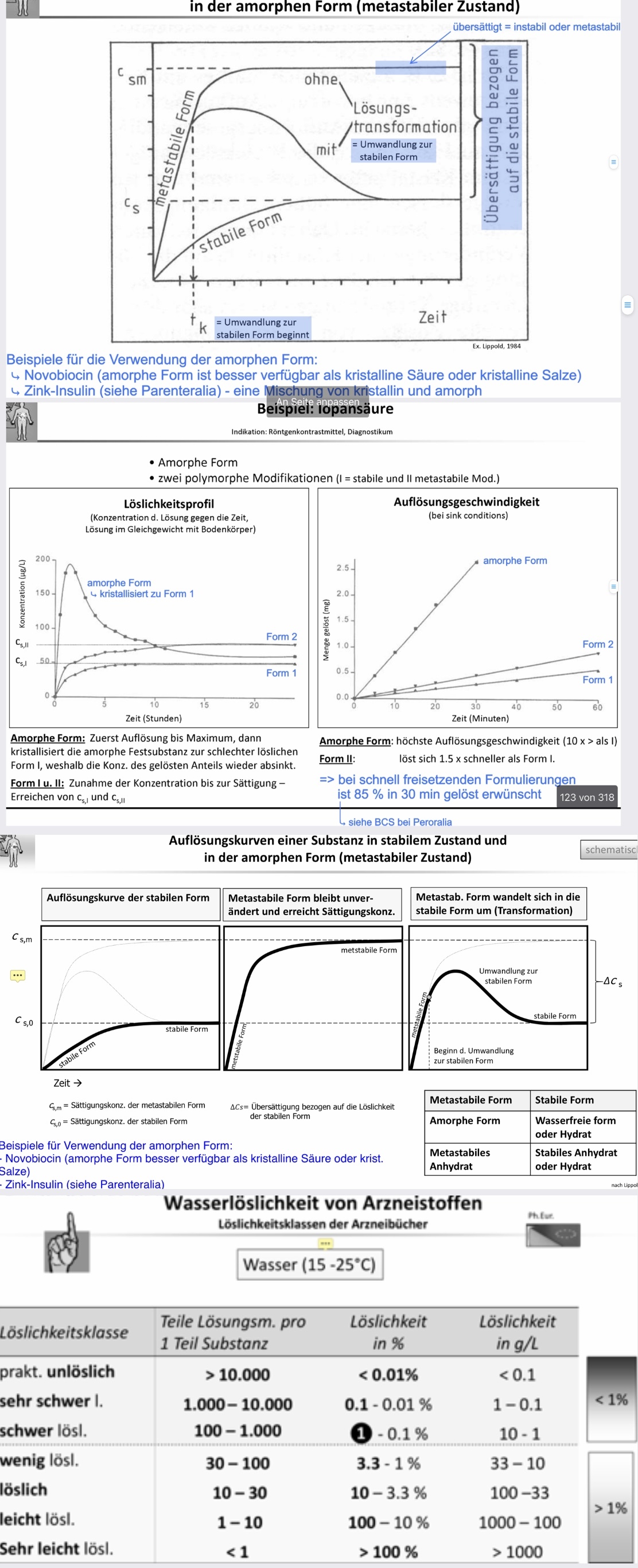
Ein Diagramm mit Kurven war gegeben, man musste zuordnen was diese Kurven zeigen
können (welche Arzneiform) und wie man die Löslichkeit bewertet
Diagramm mit Amorphe Form, Form 1 und Form 2 erklären und verhalten unter sink bedingungen zeichnen
Hier eine Abbildung mit den Auflösungskurven
In der Metastabilen Form erreichen wir eine höhere Konzentration und diese auch schneller.
Bild 1
Phasenumwandlung zu Form I - Spring Parashoot Effekt bei Amorphen Form
Bild 3
Stabile Form, erreichen wir eine niedere Konzentration und in langsamerer Geschwindigkeit
Dritte Variante ganz rechts
Lösen einen metastabilen Stoff auf aber während des Auflösens bildet sich die Stabilere Form (kann sich in der Lösung bilden - weil ab dem höchsten Punkt der Kurve ist die Lösung ja übersättigt)
–> nennt dies auch “Spring Parashoot” oder “Drachengleiter” Effekt.
Wir erreichen die Sättigungskonzentration der stabilen Form
Die Differenz (delta cs) ist eine wichtige Größe - kann ein vielfaches der Sättigungslöslichkeit der stabilen Form ausmachen. Je nachdem welche Problematik beim Wirkstoff vorliegt, kann ich mehr oder weniger Effekte erreichen.
Bewertung der Löslichkeit:
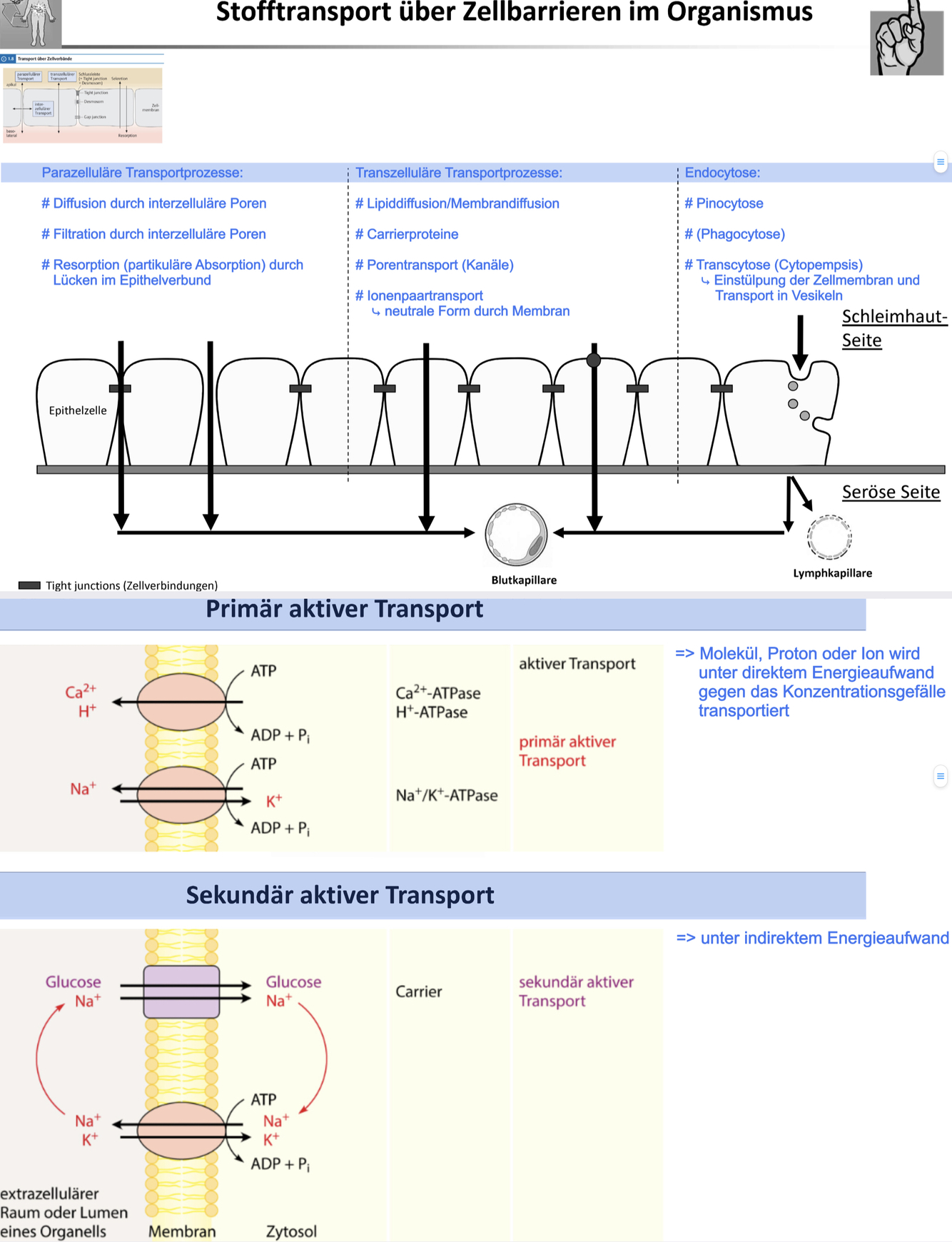
Transzelluläre Transportsysteme
Transzelluläre Transportwege 4 Stück was zeichnet sie aus zum Beispiel Größe Lipophilie?
Skizze mit Zellen beschriften:Transportprozesse (Lipiddiffusion, …)
Welche Transportmöglichkeiten von Ionen und hydrophilen Substanzen kennen Sie?
Transzelluläre Transportwege 4 Stück, was zeichnet sie aus (z.B. Größe, Lipophilie,pH )
- Lipiddiffusion (Transport von gelösten Molekülen entlang eines Konzentrationsgradienten)
- Carrier-vermittelte Transportprozsse (erleichterte Diffusion) -> ohne Energieverbrauch, entlang einen Konzentrationsgefälles spezifisch für Aminosäuren, Monosaccharid
- aktiver Transport: unter ATP-Verbrauch
- Iorentransport
Inter- und parazelluläre Transportprozesse: Diffusion
Transzelluläre Transportprozesse: Lipiddiffusion Carriervermittelte/
Vesikuläre Transporte: Pinozytose Phagozytose Endozytose
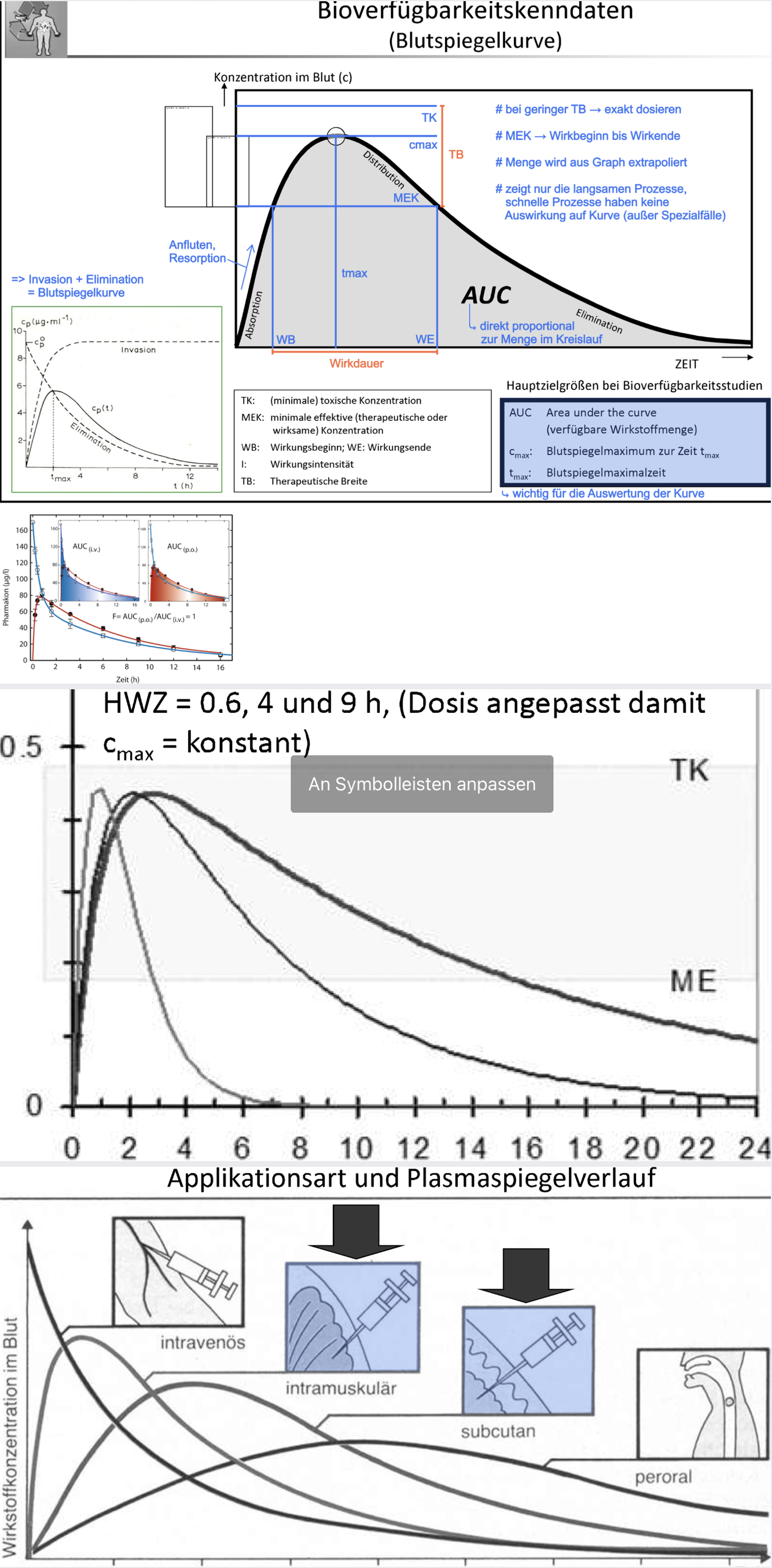
Plasmakurve zeichnen mit allen wichtigen Parametern und eine weitere Kurve einzeichnen
mit derselben Resorptionsrate aber doppelter Eliminationsgeschwindigkeit
Zeichnen Sie eine Blutspiegelkurve und deren wichtigsten Parameter/ Blutspiegelkurve und i.v. Kurve zeichnen/2 Blutspiegelkurven Absorption/Elimination
Blutspiegelkurve (peroral) -> Intravenös einfach exponentieller Abfall (keine Absorption, nur Elimination)
-Liposome, Nanopartikel, Mikropartikel > warum eine ölige Form von Vorteil sein kann?
Unterschied in der Freisetzung von öliger Lösung und Suspension (geschwindigkeitsbestimmender Schritt)
Öl wird nicht resorbiert sondern langsam abgebaut Also quasi ein (echtes) Depot, weil das Öl bleibt lang an der Injektionsstelle
Unterschied in der Freisetzung von öliger Lösung und Suspension (geschwindigkeitsbestimmender Schritt
Lösung
- Öl wird nicht resorbiert sondern langsam abgebaut Also ein echtes Depot, weil das Öl bleibt lang an der Injektionsstelle
- Stoff, der sich in einem Öl löst = hohe lipophilie Stoffe –> gehen sehr schnell durch die Membranen oder durch die Kapillaren wenn wir diesen in die Subkutis applizieren.
- Die Geschwindigkeit des Gesamtprozesses (Verteilungskoeffizient) hängt im wesentlichen davon ab, wie schnell sich dieser Wirkstoff zwischen dem Öl und der wässrigen äußeren Phase verteilt Hier haben wir “nur” die Lösung, die Lösung hat eine bestimmte Konzentration (aber wir haben kein “Resorvoir” wie bei der Suspension)
Suspension:
- Übersättigte” Lösung
- Haben einen Bodenkörper und darüber haben wir eine gesättigte Lösung, hier stellt sich ein Gleichgewicht ein
- Wenn wir jetzt aus der Überstehenden Lösung den Arzneistoff durch Diffusion in eine anderen Bereich (in die Blutkapillaren) reduzieren, dann löst sich aus dem Bereich des festen Anteils etwas nach und wir haben dadurch ein sehr gutes “Resorvoir”.
- Wenn wir diesen Wirkstoff nun auch noch in Partikulärer Form (als Kristalle) in dieses Öl einbringen, dann haben wir stärkere Verlängerungszeiten gegenüber einen reinen Lösung. Wir bilden also Suspensionen um genügend Wirkstoff in dem Öl unterzbringen

Erkläre definiere Supramolekulare Eigenschaften & Wie kann man supramolekulare Eigenschaften verändern?
Salzbildung
Amorphisieurng
Polymorphie und Pseudopolymorphie
Skizze mit Zellen beschriften:
Transportprozesse(Lipiddiffusion,..)
Ergänzung Carrier Vermittelte Transportprozesse
–> erleichterte Diffusion
–> aktiver Transport
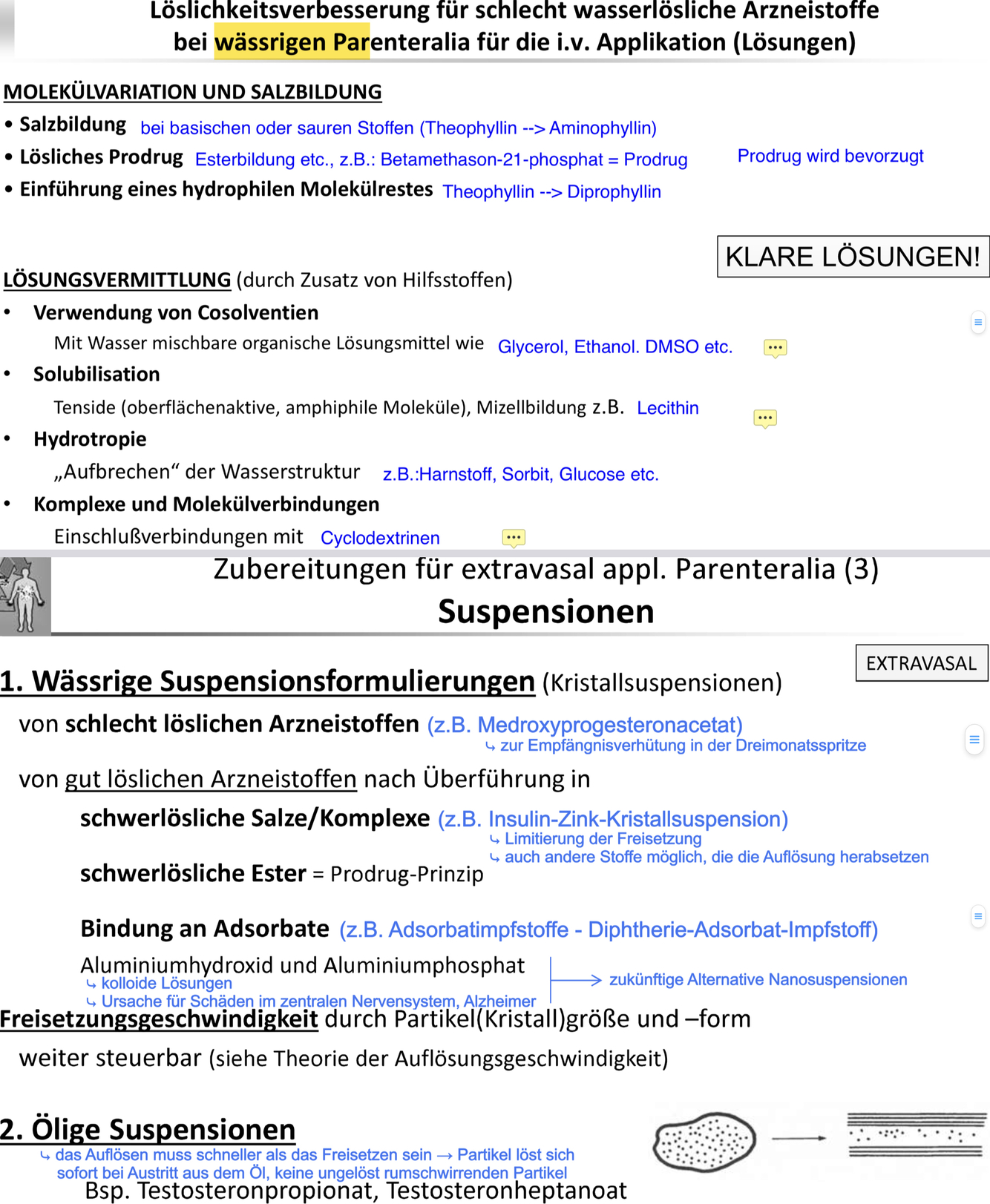
Wie kann man die Löslichkeit eines wässrigen ASt. für Infusionen verbessern?
Wässrige parenterale Injektion – wie verbessern hinsichtlich Löslichkeit?
Wie Suspension zur Injektion/Infusion für extraversal/ Parenterali beeinflussbar
welche Formulierung würden sie hier verwenden: Einzeldosis 2mg, MR iwas, chronische Krankheit, HWZ 2h -> begründe warum
Retard Formulierung, da die Einzeldosis niedrig ist und es sich um eine chronische Krankheit handelt, die dauerhal behandelt werden muss. Außerdem ist die Elimina9onshalbwertszeit relativ kurz und daher würde eine Retardformulierung, die über längere Zeit Wirkstoff freigibt/mehrere Dosen enthält Sinn machen. Man häge so auch eine bessere Compliance
Kriterien für eine Retard Formulierung; • Geringe Einzeldosis –> hier der Fall mit 2 mg
- Kurze/miglere HWZ –> hier 2h ist relativ kurz –> geeignet für Retard
- Indikation –> chronische Erkrankungen, bei denen man konstante Blutspiegel will –> hier der Fall
- Schnelle Resorption –> brauche Lipophiles, kleines Molekül
Bauprinzip/Merkmale Kristall
Aufbau/Definition Kristallstruktur
Kristall Bauprinzip/Merkmale
hochgeordnete Aggregate aus Millionen von Atomen, Molekülen oder Ionen, die sich durch gegenseitiges Erkennen selbst organisieren;
- Anisotropie
- homogener Aufbau
- 3D-periodische Ordnung der Bausteine
- Einheitszelle (7 Grundtypen, 3 Winkel, 3 Seitenlängen; meist Ast ein triklines, monoklines oder orthorhombisches Kristallsystem)
Kristallstruktur definiert durch:
- Einheitszelle
- Raumkoordinaten der atomaren Bausteine
- Symmetrie
Wie unterscheiden sich single unit dose und multi unit dose hinsichtlich
a) retardierten
b) magensaftresistenten
c) schnell freisetzende
Arzneiform in Bezug auf Einnahme und Nahrungsaufnahme?
Retardierte AF
- • Single Unit –> habe eine längere Verweildauer im Magen, da sie erst mit der Housekeeper Phase weitertransportiert wird –> gut für retardierte AF, da diese so länger im System bleiben, außerdem kann ich so das Resorptionsfenster im oberen Dünndarm gut treffen
- • Multiple Unit –> für Retard schlechter, da ich so eine schnellere Magenpassage habe und die Wirkung dadurch kürzer wird
Magensalresistente AF:
- nüchterne Einnahme sieht kaum unterschied dieser Beiden
- • Multiple Unit –> besser, da ich hier praktisch Dosis einfaach weiter transportier wird durch Magenpassage habe und dadurch die Wirkung im Darm schneller trotz essen
- • Single unit –> schlechter für magensalresistene, da es hier zu einer längeren Verweildauer im Magen kommt, was dazu fürht, dass ich erst einen sehr späten Wirkungseintrig habe
Schnell freisetzende AF in Bezug auf Einnahme und Nahrungsaufnahme
- • Multiple unit –> wird durch Nahrungsaufnahme kaum beeinflusst und kann auch mit der Nahrung eingenommen werden
- • Single unit –> durch Einnahme zusammen mit Nahrung habe ich eine wesentlich längere Transitzeit durch den Magen, was zu einer sehr stark verzögerten Wirkung und zu geringere cmax und höhere tmax führen kann
Wie könnenPeptide in Lösung gebracht und appliziert werden, beurteilen Sie dies bezogen
a) pulmonal, b) peroral c) buccal und d)nasal e)extravasal/ intravenös als Applikationsort.
Mittels Liposomen & Nanopartikel
am Bsp vo Zink-Insulin
In Lösung als Monomere oder Dimere Im kristallinen Zustand als Hexamere
Kristallinese Zink (schlecht löslichste Variante), Amoprhen Zustand (der dann eine schnellere Freisetzung und Zerfall hat)
Die Dissoziation der Hexameren zu resorbierbaren Monomeren ist der geschwindigkeitsbestimmende Schritt z.B Insulin-Zink-Kristallsuspension. –> s.c Applikation (
compliance Inhalation +++, Pumpe ++, Spritze+
Pulmonal
Sehr schnelles Anfluten, deutlich bessere Resorptionsbedingungen als bei anderen Applikationsorten (auch für Peptide ist man an erproben Insulin )
Peroral
Peptide, Proteine oder gentherapeustische Arzneistoffe wie z.B. DNA können nicht peroral verabreicht werden, weil sie im Gastrointstinaltrakt sofort abgebaut werden.
jedoch OTF (oral thin fillms) orale Haftarzneiformen / mucoadhäsion möglich
buccal
nasal
Nose to Brain delivery direkte Anbidnung an ZNS & Blut-Hirn-Schranke.
Was Peptidasen angeht gibt es gewisse Aktivität hier, die is aber so gering, dass man keine Probleme hat im vergleich zum GIT - Wenn es um die Applikation von Proteinen geht
–> geeigneter Weg für die Applikation von Proteinen (<20 AS)
extravasal/intravenös
Darreichungssformen sind injizierbar! Intravasal (Nano‐) , extravasal (mikro‐)
bioabbaubaren Polymeren erlaubt eine bessere Steuerung der Wirkstofffreisetzung wodurch das Nebenwirkungsrisiko minimiert und die therapeutische Sicherheit verbessert werden kann.
Vor- und Nachteile von peroral/extravasaler und parenteraler Applikation?
Extravasale Applikation zB orale Applikation
Vorteile:
- ✓ einfache, bequeme Verabreichung –> hohe Akzeptanz (Compliance)
- ✓ große Resorptionsfläche, gute Durchblutung –> hohe Resorptionsgeschwindigkeit
- ✓ Verweildauer von mehreren Stunden ! Anwendung von Depotarzneiformen und Af mit kontrollierter Freisetzung möglich
- ✓ geringe Herstellungskosten
Nachteile:
- ✓ First-Pass-Effekt und Efflux-Mechanismen
- ✓ langsamerer Wirkungseintritt, da zunächst Resorptionsschritt (Aufnahme ins Blut) nötig ist.
- ✓ Resorptionsschwankungen wegen:
- ✓ variablen Resorptionsbedingungen (zB pH Magen/Darm)
- ✓ pathologische Zustände im GIT (zB Malabsorption)
- ✓ WW mit versch. Ast. ✓ limitierte Applizierbarkeit von Ast. wegen:
- ✓ hoher metabolischer/enzymatischer Aktivität (zB Abbau von Peptiden, zB Somatotropin)
- ✓ extremen pH-Werten (zB Abbau von säurelabilen Stoffen im Magen)
- ✓ schwer überwindbare Epithel im GIT (zB Resorptionsbarriere für hydrophile Makromoleküle wie Peptide, Proteine etc.)
- ✓ Irritation oder Schädigung der Schleimhaut durch gastrotoxische Wirkstoffe möglich
Parenterale Applikation
Vorteile:
- ✓ gezielte Applikation des Ast in bestimmtem Gewebe
- ✓ kontrollierte Verabreichung, präzise Dosis
- ✓ Umgehung des Magens und der ersten Leberpassage (auch Peptide können appliziert werden)
- ✓ Therapie trotz Resorptionsstörung möglich
- ✓ rascher Wirkungseintritt (intravasal, Akutbehandlung)
- ✓ Behandlung von bewusstlosen Patienten möglich (Notfälle)
- ✓ Anlegen von Langzeitdepots bei extravasaler Applikation
Nachteile:
- ✓ hohe Anfangskonzentrationen -> lokale Unverträglichkeiten an der Injektionsstelle
- ✓ kein Abbruch möglich bei Überdosierung, Überreaktion
- ✓ Infektionsrisiko an Einstichstelle
- ✓ Gefahr bei versehentlicher paravasaler Applikation
- ✓ hoher Qualitätsstandard der Arzneiform nötig
- ✓ Abhängigkeit von befugten Personen
- ✓ Compliance Probleme (Schmerz)
.Parameter gegeben, Beurteilung welche Applikation dafür die beste wäre (Akuttherapie,max. Einzeldosis 5mg, hohes MG, hydrophil, Löslichkeit von 30mg/L…)
Akkute iv. oder p.o –> Nüchtern einnehmen !!!
EMD–> klein spricht für Nasal, Buccal, sublingual, Pulmonal
Resorption –> eher unggeschickt das groß & hydrophil
Das Molekül ist sehr groß (Großes Molekulargewicht) wird oral nicht resorbiert
Löslichkeit– 0,033g/L < 0,1g/L = prakt. unlöslich
Fazit
Nasal or pulmonal
Als Liposomen Infusionszubereitung –> i.v jedoch da akkute würd ich die oberen beiden beverzugen, für das lipsoem spricht großes Mr & hydrophil…
Vesikel eingelagert und kann relativ gut appliziert werden. Der Einschluss in dem Lipophilen Bereich bedeutet auch, dass der Wirkstoff nicht frei gelöst im Blut vorliegt sondern assoziiert in diesem Phospholipid Bereich der Partike
